
译者:淡月疏星 公众号:药有源

伏硫西汀(Vortioxetine)由灵北和武田制药联合研发,2013年9月获FDA批准上市,用于成人抑郁症(MDD)的治疗,同年12月获EMA批准,剂型为口服薄膜包衣片和滴剂,商品名“TRINTELLIX”。因其为具有临床价值的新药获CDE优先审评,其片剂于2017年在国内批准上市,商品名为“心达悦”。互联网信息显示,国内已有3家企业提请仿制药申请。


2.2.2 原料药
伏硫西汀(Vortioxetine)已证实为新活性成分,采用伏硫西汀氢溴酸盐和伏硫西汀DL-乳酸盐分别制备了速释薄膜包衣片和口服滴剂。
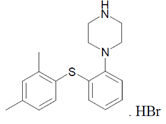
氢溴酸伏硫西汀(Vortioxetine hydrobromide)
氢溴酸伏硫西汀的化学名称为1-[2-(2,4-二甲基-苯基磺胺基)-苯基]-哌嗪氢溴酸盐,其结构如下:
生产
伏硫西汀氢溴酸盐通过两个明确定义的合成步骤生产,然后进行重结晶和研磨。所使用的起始原料定义明确,并且从可接受的合格供应商获得。伏硫西汀氢溴酸盐的生产涉及两个生产场所。已经充分详细地描述了合成工艺并进行了充分的过程控制。明确了中间产品、起始原料和试剂的质量标准和控制方法。充分地讨论了生产过程中杂质的形成、存在、来源和清除过程。活性物质及其杂质的表征符合欧盟关于新活性物质的指南。关于潜在杂质和实际存在杂质的来源和特性进行了充分讨论。提供的两个拟定的生产场所的代表性批次分析数据显示,采用拟定的合成工艺可重复生产出符合要求的活性物质。
质量标准
原料药的质量标准包括鉴别(HPLC、FTIR和NIR)、含量测定(HPLC)、有关物质(HPLC和GC)、残留溶剂(GC)、炽灼残渣、重金属和粒径。详细描述了分析方法,非药典分析方法已根据ICH指南进行了充分验证。提供了多批商业批和开发批次原料药的批分析数据,均符合质量标准且批间一致性良好。
稳定性
提供了按申报工艺生产并采用拟上市包装的2批中试批和9批商业批氢溴酸伏硫西汀原料药的稳定性数据。根据ICH指南提供了上述批次长期(25℃/60%RH)48个月和加速(40℃/75%RH)6个月的稳定性数据。采用1批原料药根据ICH Q1B进行了光稳定性研究,并采用1批原料药在60℃/80%RH条件下进行了强制降解试验。对以下属性进行了考察:性状、含量和有关物质。所采用的分析方法同放行标准且可以反应稳定性的变化。稳定性数据表明采用申报工艺生产的原料药稳定,且证明拟定包装中的复验期是合理的。
伏硫西汀DL-乳酸盐(Vortioxetine DL-lactate)
伏硫西汀DL-乳酸盐化学名称为1-[2-(2,4-二甲基-苯基硫烷基)-苯基]-哌嗪(RS)-2-羟基丙酸酯,并具有以下结构:
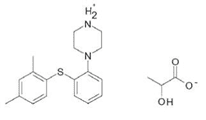
分子式为C18H22N2S.C3H6O3,相对摩尔质量为388.52mg/mol。由于乳酸盐在极性溶剂中较好的溶解度而用于滴剂。伏硫西汀乳酸盐为白色至米色粉末,无吸湿性,溶于极性溶剂和水,游离碱pKa为9.1、盐为3.0。伏硫西汀具有3种多晶型。已经确定了热力学上最稳定的晶型,并且设计了结晶过程以确保始终获得这种晶型。
生产
伏硫西汀DL-乳酸盐是由经研磨的氢溴酸伏硫西汀通过非分离的游离碱制得的。伏硫西汀乳酸盐的生产在一个生产场所进行。充分详细地描述了合成过程及过程控制措施。提出了中间产品、起始原料和试剂的质量标准和控制方法。充分地讨论了生产过程中杂质的形成、存在、来源和清除过程。活性物质及其杂质的表征符合欧盟关于新活性物质的指南。关于潜在杂质和实际存在杂质的来源和特性进行了充分讨论。提供的拟定的生产场所的代表性批次分析数据显示,采用拟定的合成工艺可重复生产出符合要求的活性物质。
质量标准
原料药的质量标准包括鉴别(HPLC、FTIR)、含量测定(HPLC)、有关物质(HPLC和GC)、残留溶剂(GC)、灼烧残渣、重金属和粒径。详细描述了分析方法,非药典分析方法已根据ICH指南进行了充分验证。提供了多批商业批和开发批次原料药的批分析数据,均符合质量标准且批间一致性良好。
稳定性
提供了按申报工艺生产并采用拟上市包装的3批中试批伏硫西汀乳酸盐原料药的稳定性数据。根据ICH指南提供了上述批次长期(25℃/60%RH)48个月和加速(40℃/75%RH)6个月的稳定性数据。采用1批原料药根据ICH Q1B进行了光稳定性研究,并采用1批原料药在60℃/80%RH条件下进行了强制降解试验。对以下属性进行了考察:性状、含量和有关物质。所采用的分析方法同放行标准且可以反应稳定性的变化。稳定性数据表明采用申报工艺生产的原料药稳定,且证明拟定包装中的复验期是合理的。
2.2.3 制剂
薄膜包衣片
制剂开发
伏硫西汀速释薄膜包衣片包含5、10、15或20mg伏硫西汀(氢溴酸盐)。制剂开发过程中,申请人对不同处方进行了评估以筛选出最适宜的辅料组成,以及理化和生物学性质、片剂属性和溶出度最佳的处方。制剂的溶出度受粒径影响,因此对原料药进行了研磨以在混合工艺中获得适宜的粒度分布。通过二元混合试验证明原辅料相容性良好。所有辅料均为药用成分并符合欧洲药典要求。制剂中未使用新辅料。所用辅料清单列在说明书6.1部分。临床试验中使用的处方与申报上市处方相同。5mg、10mg、15mg和20mg薄膜包衣片工艺开发的中试批样品采用与生产规模相似的设备生产。薄膜包衣片采用PVC/PVdC铝塑泡罩和HDPE包装。包装材料符合欧洲药典和EC要求。通过稳定性研究说明了容器包装系统的选择依据,并与产品预期用途相符。
外来物质
未使用动物源或人源辅料。
制剂生产
该速释制剂的生产过程包括以下步骤:混合、流化床制粒、干燥、混合、压片和包衣。该生产过程可认为是标准生产过程。主要生产步骤经过一系列研究进行了验证。已证明生产过程可持续生产出符合质量要求的制剂。针对该生产工艺进行了充分的生产过程控制。
制剂质量标准
制剂放行标准包括此类剂型的检查项,包括性状(目视)、鉴别(HPLC、UV和FT-IR)、含量均匀度、含量(HPLC)、降解产物(HPLC)、溶出度、水分和微生物限度(Ph. Eur.)。详细描述了分析方法,非药典分析方法已根据ICH指南进行了充分验证。提供了每个规格3个中试批产品的批分析结果,证明了生产工艺的一致性和可生产出符合预期质量标准产品的稳定性。
制剂稳定性
根据ICH指南,提供了3批中试规模制剂长期条件(25℃/60%RH)和中间条件(30℃/75%RH)24个月,以及加速条件(40℃/75%RH)6个月的稳定性数据。进行稳定性研究的制剂批次可以代表拟上市产品并采用拟上市初级包装。考察了片剂的性状、含量、降解产物、溶出度、水分和微生物限度。所采用的分析方法可以反应稳定性的变化。此外,采用1批制剂根据ICH新原料药和制剂光稳定性指南进行的光暴露研究,并在25℃/93%RH条件下进行了3个月的高湿考察。根据提供的稳定性数据,产品使用说明书中规定的有效期和贮藏条件是可接受的。
口服滴剂
制剂开发
预期目标为开发20mg/ml的口服滴剂且采用滴管给药20滴时剂量为20mg伏硫西汀。与薄膜包衣片采用氢溴酸盐作为活性成分不同,口服滴剂采用DL-乳酸盐作为活性成分,该活性成分在极性溶剂中具有更高的溶解度。制剂处方中的羟丙基-β-环糊精进一步提高了原料药的溶解度,在开发过程中考察了该辅料的取代度及其与活性成分相互作用所需温度,两个参数对羟丙基-β-环糊精和伏硫西汀的相互作用均不关键。另一辅料96%乙醇增加了每毫升溶液的滴数并同时作为防腐剂。在8.5%(w/v)浓度下考察了不同比例用量并获得预期的20滴/毫升。通过抑菌效力试验证明口服滴剂的处方自身具有防腐作用。纯化水为该处方的溶剂。所有辅料均为药用成分并符合欧洲药典要求。制剂中未使用新辅料。所用辅料清单列在说明书6.1部分。临床生物等效性试验所用的处方与申报上市处方相同,其结果证明20mg/ml口服滴剂溶液与20mg规格薄膜包衣片等效。口服滴剂初级包装为安瓿玻璃瓶和具有防儿童开启功能的螺旋盖。包装材料符合欧洲药典和EC要求。通过稳定性研究说明了容器包装系统的选择依据,并与产品预期用途相符。
外来物质
未使用动物源或人源辅料。
制剂生产
生产工艺包括三个主要步骤:配液、过滤和安瓿瓶灌装。生产过程需要避光。该生产过程可认为是标准生产过程。主要生产步骤经过一系列研究进行了验证。已证明生产过程可持续生产出符合质量要求制剂。针对该生产工艺进行了充分的生产过程控制。
制剂质量标准
制剂放行标准包括此类剂型的检查项,包括性状(目视)、鉴别(HPLC、UV)、伏硫西汀含量(HPLC)、96%乙醇(GC)、降解产物(HPLC)、口服滴剂的剂量和剂量均一性、pH(Ph. Eur.)和微生物限度(Ph. Eur.)。详细描述了分析方法,非药典分析方法已根据ICH指南进行了充分验证。提供了3个批次产品的批分析结果,证明了生产工艺的一致性和可生产出符合预期质量标准产品的稳定性。
制剂稳定性
根据ICH指南,提供了2批商业生产规模和1批中试规模制剂长期条件(25℃/60%RH)和中间条件(30℃/75%RH)18个月,以及加速条件(40℃/75%RH)6个月的稳定性数据。进行稳定性研究的制剂批次可以代表拟上市产品并采用拟上市初级包装。考察了制剂的性状、伏硫西汀含量、乙醇含量、降解产物、口服滴剂的剂量和剂量均一性、pH、溶液颜色和微生物限度。所采用的分析方法可以反应稳定性的变化。此外,采用1批制剂根据ICH新原料药和制剂光稳定性指南进行的光暴露研究,并采用2批制剂在药瓶首次开启后在25℃/60%RH条件下进行了长达8周使用中稳定性研究。根据提供的稳定性数据,产品使用说明书中规定的有效期和贮藏条件是可接受的。
2.2.4 化学、药剂学和生物学讨论
关于原料药和制剂的开发、生产和控制的资料已经以令人满意的方式提交。提供的试验结果表明产品关键质量属性的一致性和均匀性,进而证明该产品在临床应用中具有令人满意且均一的性能。
2.2.4 化学、药剂学和生物学结论
当按照产品使用说明书中规定的条件使用时,本产品的质量被认为是可接受的。已对与产品临床性能相关的物理化学和生物学方面进行了研究,并以令人满意的方式进行了控制。
© 版权声明
文章版权归作者所有,未经允许请勿转载。
相关文章



暂无评论...