
来源:药学有源
导读
EMA审评报告原文翻译
概述
Sarclisa为供输注使用的浓缩液,含有20mg/mL的活性成分isatuximab,两种瓶装规格100mg/5mL和500mg/25mL。其他成分为:组氨酸,组氨酸盐酸盐一水合物,蔗糖,聚山梨酯80和注射用水(WFI)。
原料药
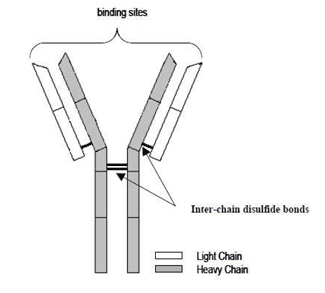
Isatuximab是在中国仓鼠卵巢(CHO)细胞株中表达的嵌合免疫球蛋白IgG(IgG1)的衍生化单克隆抗体(mAb)。Isatuximab与分化簇38(CD38)受体的特定细胞外表位结合,引发多种机制导致表达CD38的肿瘤细胞死亡。蛋白质结构(图1)由两条分子量约为23kDa的κ轻链和两条分子量约为49kDa(去糖基化形式)的IgG1重链通过二硫键连接而成。每条轻链由214个氨基酸残基组成,每条重链由450个氨基酸残基组成。重链N末端谷氨酰胺残基被完全转化为焦谷氨酸。大部分重链C末端K450被剪切(使用缺失肽图分析C末端K450在9至11%之间)。Isatuximab包含32个半胱氨酸,形成16个二硫键,并含有位于重链天冬酰胺N300上的两个糖基化位点。
原料药生产、过程控制和表征
生产
申请人提供了制造商和合同实验室执行生产、储存和控制isatuximab原料药(formulated drug substance,FDS)活动的描述,提供的资料是可以接受的。
生产过程描述和过程控制
用于2期和3期临床试验以及过程性能确认(Process Performance Qualification,PPQ)的FDS原材料,代表了商业生产过程。活性成分的生产过程始于工作细胞库(WCB)的解冻、细胞培养和CHO细胞的收获。该过程包括以下步骤:在摇瓶和波浪袋中扩增培养物,然后通过扩增生物反应器,以接种10,000 L的生产生物反应器生产并最终收获isatuximab抗体。最后通过一系列色谱、过滤和病毒灭活步骤实现isatuximab的纯化。这一系统的纯化步骤旨在去除生产和与产品相关的杂质,有效获得isatuximab蛋白,浓缩并将药物物质交换到最终的缓冲系统中。对生产过程进行了充分的描述和证明,并提供了足够的详细信息,包括每个生产步骤的过程控制和工艺参数。提供了isatuximab FDS生产过程保持时间验证(生产的最长时间限制)。这些资料都是可接受的。
物料控制
已列出所有使用的物料,并提供了非药典物料的质量标准。除了CHO生产细胞株外,生产过程中没有使用任何生物来源的物料。表达isatuximab的中国仓鼠卵巢(CHO)宿主细胞株是无血清悬浮细胞株。针对细胞株的培养和细胞库的建立也作了足够详细的描述。根据ICH Q5A和ICH Q5D,对主细胞库(MCB)、工作细胞库(WCB)和达到体外年龄极限的细胞(LIVCA)进行了鉴定,并进行了无菌、支原体检测、同工酶分析、HC-和LC-链基因序列和侧翼区的控制以及病毒检测,均符合验收标准;表明细胞库来源于仓鼠,具有正确的基因序列,无菌,除了逆转录病毒样颗粒外未检测到支原体和病毒,符合对CHO细胞的预期。还提供了一份今后可能建立WCB的详细可接受标准的规程。这个协议被认为是可以接受的。此外,还提供了确认生产细胞株遗传稳定性的可接受数据。
关键控制步骤和中间体
已经为isatuximab活性成分生产中的每个过程步骤定义并总结了过程控制(关键或非关键IPC)和工艺参数(CPP或非CPP)。对过程控制和工艺参数的可接受范围进行分级和论证的基本原由进行了详细和可接受的论证。过程控制(关键和非关键IPC)和工艺参数(CPP或非CPP)都包含在申报资料的此部分。应当指出,关键和非关键IPCs和CPPs或非CPPs在今后的任何变更都通过变更申请处理。IPCs的可接受标准和行动限根据生产经验和先前的知识确定。已经为细胞培养、收获、纯化和配方的生产步骤建立了安全相关的IPCs。这些IPCs包括生物负荷、细菌内毒素、支原体和病毒检测。除了IPCs,质量标准中还针对活性成分的批放行制订了生物负荷和细菌内毒素检查。关键步骤和中间体控制部分提供的信息是可以接受的。
工艺验证和/或评价
Isatuximab FDS的商业生产过程已经过验证,证明当在规定的工艺参数内运行时,该生产过程能够满足预先设定的可接受标准。工艺验证和评价数据是全面的,支持生产工艺的持续性。提供了用于评价生产工艺以及确定关键质量属性(CQAs)和CPPs的工艺参数和质量属性的可接受范围。验证和评价是在几个工艺性能确认(PPQ)批次上进行的。提供并总结了四个PPQ批次的每个生产步骤的关键和非关键工艺参数以及质量属性的结果。PPQ批次符合所有质量标准的可可接受标准,证明对产品和工艺相关的杂质的清除率始终保持一致。工艺验证PPQ批次在商业规模下进行,但某些方面的验证以缩小的规模进行,例如病毒清除率和树脂寿命研究。缩小规模的研究使用经认定的按比例缩小模型。对活性成份运输到制剂生产场所的过程进行了验证了并证明运输过程有效。提供的有关工艺验证的信息是可以接受的。
生产过程开发
全面描述了生产工艺开发过程,包括工艺开发和开发过程中的变更以及对比研究。此外,还提供了商业生产过程表征、CQAs的确定以及有关容器密封系统适用性的部分信息。对过程控制(关键或非关键IPC)和工艺参数(CPP或非CPP)的可接受范围进行分级和论证的基本原则进行了详细论证。
工艺开发过程及其变更
工艺开发历史已被详细描述。Isatuximab活性成分生产的预期商业工艺已被用于生产临床II期和III期研究用的活性成分。在不同的开发阶段均使用的相同的细胞株。研究了开发过程中工艺的可比性。根据所提供的结果,不同工艺生产的活性成分整体表现出相当的特征,在可比性部分提供的信息被认为是充分合理和可接受的。
控制策略
充分描述了过程控制策略,包括针对商业生产工艺在多个开发阶段的一系列系统研究。通过多学科风险评估确定了关键控制属性,包括对最终制剂安全性和/或有效性的影响。提供了所有CQAs和非CQAs。
商业生产中的过程表征
最初的风险评估源于先前的认知、研发和生产历史。过程表征研究包括工艺步骤小规模模型和建立的经证明的可接受范围(PARs)。过程表征的结果也分别用于定义工艺的CPPs和非CPPs,以及CPPs和非CPPs的可接受标准和行动限。经证明的可接受范围基于实验设计(DOE)方法模型确定。成功生产了工艺性能确认(PPQ)批次证明生产工艺的可重现性。总结了所有最终确定的CPPs和非CPPs。提供了CPPs确定的依据。采用多变量理解来增加对工艺认知并建立工艺参数范围的策略是可行的。在这种情况下,提供了足够的数据并且所提出的工艺参数是固定的,因此可以得出结论,对于isatuximab生产工艺步骤提出的经证实的可接受范围(PAR)是合理和可接受的。提供的控制策略、CQAs的确定和商业生产工艺中的表征研究的信息被认为是可接受的。
表征
采用最新的方法对isatuximab活性成分进行了全面的表征。所有表征研究采用代表商业生产工艺的isatuximab进行。
杂质
已对活性成分潜在的工艺相关杂质和产品相关杂质进行了检测。所有杂质均认为可通过生产工艺、推荐的贮藏条件和相关的分析监控得到良好控制。
质量标准、分析方法、标准物质、批分析和包装
活性成分的质量标准包括外观、鉴别、效力、纯度、细菌内毒素。申请的测试和标准限度是可接受的。除了某些仅在放行时执行的检测外,可接受标准对放行和货架期产品均有效。这是可以接受的。活性成分质量标准中的可接受标准是基于商业生产工艺表征结果和安全性数据的生产能力确定的。所有与生产过程和产品相关的杂质都认为可以通过生产、推荐的贮藏条件和相关的分析监控得到良好的控制,因为在商业规模的生产过程中,所有这些活性成分的杂质均已被证明可以降到很低或未检出的水平。
分析方法
提供用于活性成分检测的分析方法,方法描述可接受且足够详细。提供了方法验证资料包括相关的转移验证,能够证明方法是恰当的。此外,还提供了isatuximab活性成分微生物限度和细菌内毒素检查适用于药典方法的报告。
批分析
提供了isatuximab活性成分开发过程中生产的所有批次的批分析数据。所有数据均符合申请的活性成分质量标准。提供了所有PPQ批次的分析结果。总结了分析方法开发过程和方法对比桥接研究结果。所提供的信息是充分和可接受的。
标准物质
建立了内部使用的一级和工作标准物质。现在两种标准物质均源自根据拟定商业工艺生产的一个批次。标准物质已经被很好地描述和表征,可代表商业生产过程。
包装容器密封系统
所选择的包装容器密封系统是充分的,符合相关标准,并已进行了足够详细的描述。
稳定性
提供了加速和苛刻贮藏条件下的稳定性数据以及光稳定性数据。稳定性研究的所有批次均能代表商业生产过程。在长期贮藏条件下,报告的稳定性数据中的任何控制参数都没有下降的趋势。根据长期、加速稳定性和破坏试验结果,提出的isatuximab活性成分的有效期是可接受的。
制剂
产品描述和制剂开发
制剂成品(FP)为用于输注的无菌浓缩液,含有活性成分isatuximab20mg/mL。其他成分包括组氨酸,盐酸组氨酸一水合物,蔗糖,聚山梨醇酯80和注射用水(WFI)。没有使用新赋形剂。Sarclisa成品包括两种规格:500 mg/25 mL和100 mg/5 mL:
•100 mg/5mL产品装于6mL一次性玻璃瓶中,包装形式为1瓶或3瓶。研究确定灌装体积(5.4mL)以确保可抽取5mL。
•500 mg/25mL产品装于30mL一次性玻璃瓶中,包装形式为1瓶。研究确定灌装体积(26.0mL)以确保可抽取25mL。
处方开发
递交了处方开发充分的资料,确定的处方合理。活性成分和辅料均没有过量投料,过量灌装是合理可接受的。
生产工艺开发
对生产工艺进行了充分的研究、描述和讨论。所选择的开发方法是恰当的。实验室规模和工业化规模的过程表征研究以及不同阶段的风险评估都是可接受的,并证明最终确定的CQAs、CPPs和控制策略是合理的。设备中可提取物和可浸出物的风险已得到可接受的解决。没有发现任何组分对潜在的可提取物和可浸出物构成高风险。此外,补充的过滤器评估证明了所选无菌过滤器的适用性。选择的初级包装材料的合理性已证明。初级包装材料(I型无色透明玻璃小瓶和I型涂层橡胶塞)符合Ph.Eur的药典要求。可提取物和可浸出物评估的结果表明,没有与有机化合物和元素杂质相关的问题。
相容性
研究包括了临床上使用的不同材料和泵组合。结果证实,经0.9%氯化钠和5%葡萄糖中稀释后,在5℃下存放48小时后再在室温下存放8小时,物理化学稳定性是可接受的。此外,在欧洲药典条件下进行了挑战试验,评估了输注溶液的抑菌性和抗真菌性。SmPC第6.3节中的表述与EMA GL CPMP/QWP/159/96“关于首次打开或复溶后人用无菌产品最大保存时限的指导原则”中的表述一致。如延长使用中的存储时间(2-8℃24小时)至比指南规定的更长,则由使用者负责。
生产工艺和过程控制
生产过程和过程控制已经在流程图中充分地描述和总结。简而言之,生产过程包括以下步骤:活性成分isatuximab(FDS)解冻、集中FDS并均质化、溶液预过滤、除菌过滤,无菌灌装、检查以及最终的标签和包装。没有再处理。
关键步骤和中间体控制
根据生产工艺开发的结论,对所选择的控制策略进行了适当的描述。最终的CQAs、CPPs和生产控制策略被认为是合理的。此外,CPPs和非CPPs的设定值和范围被认为是合理的。所列关键工艺参数在最终风险评估中进行了评估。明确了CPPs和非CPPs分级的合理性。
工艺验证和/或评价
采用传统方法进行工艺验证,包括每个规格(100 mg/5 mL和500 mg/25 mL)三个连续生产规模批次。验证包括两个规格不同的批体积以及常规和最长操作时间,包括保持时间和允许的最大不制冷时间(TOR)。所有验证结果均符合验收标准,且在使用常规或最长操作和保持时间(也包括计划的工艺中断)制造的批次之间没有观察到差异。在PPQ过程中,对整个生产过程的不制冷时间(TOR)进行监控。生产完成后对加工时间最长的批次进行额外的非制冷时间研究。这些样品进行了稳定性考察且可用数据符合质量标准。
总之,工艺验证是可接受的,证明成品符合验收标准和质量标准,并能在规定的生产和保持时间内制造出一致的产品。
过滤验证和培养基模拟灌装
通过物理化学性质和培养基模拟灌装证明预过滤溶液的最大保持时间是可接受的。提供了预滤器和无菌滤器的可接受信息,包括材料、孔径、表面积和灭菌工艺。过滤器验证研究中,对过滤器的完整性、化学相容性、颗粒释放试验、细菌挑战试验和生存试验、吸附、pH值和可提取物进行了研究。验证研究的参数反映了过滤工艺的最坏情况。结果表明所选过滤器适用于预过滤和除菌过滤。养基模拟灌装试验包括预过滤后的保持时间、产品散装溶液的过滤和成品溶液的灌装。结果显示没有阳性样品,说明确定最大过滤时间是合理的。
运输验证研究
这项研究是为了考察运输过程的影响。模拟运输验证后的成品符合可接受标准。经验证两个规格产品的包装结构(100 mg/5mL和500 mg/25 mL)均适用于商业化产品。
质量标准、分析方法和批分析
质量标准
成品质量标准包括外观、鉴别、效价、纯度、无菌、细菌内毒素和可抽取体积。拟定的检查项目和可接受标准是可接受的。确定的质量标准符合欧洲药典人用单克隆抗体通则要求。此外,无菌、细菌内毒素、灌装量和颗粒物检查均符合欧洲药典0520关于肠外注射液的要求。
杂质
对潜在的工艺相关杂质和产品相关物质进行了充分的阐述,并针对每个杂质制定了控制策略。元素杂质按照ICH Q3D指南的要求进行监测。提供了风险评估总结。研究过程中,没有检测到超过分析评估阈值的有机化合物,也没有检测到超出非肠道化合物ICH Q3D限度的元素杂质。
分析方法
根据ICH Q2对分析方法进行了验证。验证结果符合要求,表明分析方法适合其预期用途并有效。
批分析
提供了商业P2F2工艺(500 mg/25mL和100 mg/5mL)的批分析数据。几个P2F2批结果涵盖了主要稳定性和临床批次以及PPQ批次,这些结果也用于正在进行的验证性稳定性研究。PPQ批次符合可接受标准并证明批间一致性符合要求。这也适用于提供的额外的批结果(包括主要稳定性和临床批次)。
标准物质
与活性成分使用相同的标准物质。可接受。
容器密封系统
Sarclisa成品装于以ETFE(乙烯和四氟乙烯共聚物)镀膜的溴化丁基塞密封的I型玻璃瓶中,铝盖密封。已确定的灌装体积(分别为5.4ml和26ml)可确保可抽取体积为5ml和25ml。
稳定性
成品申请有效期为36个月,避光贮藏储存于2-8℃。提供了按商业工艺生产批次和中试批次的稳定性研究结果。这些研究根据ICH Q5C(生物技术/生物制品的稳定性测试)进行。提供了加速和破坏试验研究结果。
主要稳定性批
5℃考察36个月所有结果均符合可接受标准。
PPQ批的验证性稳定性研究
PPQ批采用与主要稳定性批次相同的条件进行稳定性试验,还包括每个规格一个批次的光稳定性试验。PPQ批次的结果和主要稳定性批次的趋势相似。光稳定性研究证实了未开封的小瓶需要避光。
结论
申请的两种包装规格在2-8℃下36个月的有效期是可以接受的。Sarclisa经稀释后在2-8℃存放48小时后再室温存放8小时(包括输液时间),研究证明了其使用中的物理化学稳定性。微生物方面,该产品应立即使用。如不立即使用,使用前的存放时间和贮藏条件由使用者负责,通常在2-8℃下不超过24小时,除非在受控和经验证的无菌条件下进行稀释。
外来物质
除了CHO细胞外,isatuximab的生产过程中没有直接使用动物源或人源的材料。已提供了信息并确认生产过程符合EMA/410/01第3版指南,将TSE传播风险降至最低。提供了有关细菌、真菌和支原体的控制和检测资料。也提供了MCB、WCB和体外年龄极限的细胞(LIVCA)检测方面的信息。总之,根据ICH Q5A和Q5D进行了适当的测试试验。作为过程控制,对每批产品未加工原液(UPB)进行外来因子检测。所进行的检测是充分和可接受的。另提供了几批UPB的数据,没有检测到外来因子,只有CHO细胞引入的逆转录病毒样颗粒(RVLP)。
病毒清除研究
有关病毒清除研究的信息是可以接受的。病毒清除研究的设计符合ICH Q5A的要求,并使用了相关的模型病毒。实验选择了最差条件。提供了有关病毒清除测试步骤缩小规模的详细信息。定量病毒风险评估表明,生产过程中逆转录病毒具有显著的安全边际。所提供的数据表明,通过对起始原料和物料的控制,对未加工的原液中的病毒污染充分的IPC,以及在isatuximab的生产过程中有效地减少广谱病毒的结果,从而确保了产品在外来因子因素污染方面的安全性。
化学、药学和生物学方面的讨论
上市许可申请中提供Sarclisa整体质量文件是充分的。在审评过程中没有重大反对意见。正如在审评过程中所讨论和明确的,转录后修饰的确定将包括在今后活性成分和制剂生产过程变理可比性研究和稳定性研究中。已经提供了充分的关于活性成分和制剂的开发、生产和控制的信息。已有良好的控制策略来保证成品质量的一致性。检测结果表明活性成分和制剂的生产工艺是经过验证且能够良好控制的。根据SmPC中定义的使用条件,Sarclisa的整体质量是可接受的。
化学、药学和生物学方面的结论
综上,基于对所提供数据的审评,从质量的角度来看,可以批准Sarclisa的市场营销许可申请。
对后续质量开发的建议
N/A
© 版权声明
文章版权归作者所有,未经允许请勿转载。
相关文章


暂无评论...