来源:药学有源
导读
盐酸右美托咪定是目前临床主流的麻醉用药。Orion Corporation于2010年9月29日通过集中程序向EMA提交了盐酸右美托咪定(Dexdor)的上市申请,因其为重大医疗创新获得集中程序资格,2011年9月16日获得批准,用于18岁以上在插管期间或插管后监护中需要轻至中度镇静的患者。该产品已作为参比制剂经国家药品监督管理局公布。本品节选其审评报告中质量部分。注:欧盟的药品上市许可申请程序包括集中程序(CP)、分权程序(DCP)和互认程序(MRP)、成员国程序(INP),其中集中程序(centralised procedure)是直接向EMA提出申请,一经批准上市整个欧盟地区有效。对于生物技术药,先进的治疗手段,罕见病用药,用于治疗艾滋病、癌症、神经退行性疾病、糖尿病、病毒性疾病、自身免疫疾病和其他免疫缺陷疾病的新活性成分的药品等,必须通过集中程序审批。2.2.1.概述
Dexdor是用于输液的浓缩溶液,含有118µg /ml的盐酸右美托咪定,相当于100µg /ml的右美托咪定。该产品有三个规格:200µg /2ml安瓿、400µg /4ml和1000µg /10ml单次使用西林瓶。2ml标示灌装量规格的内包材为I型无色玻璃安瓿,另外两个规格(4 ml和10 ml标示灌装量)为I型无色玻璃瓶和两种可替换使用的涂有含氟聚合物膜的溴化丁基橡胶塞(Omniflex plus和ETFE),并采用用铝盖和聚丙烯翻盖密封。Dexdor只能经稀释后采用受控的输液设备经静脉输液使用。研究了可相容性的稀释液和给药装置/袋,研究确定了稀释后输液的使用时间和贮存条件。Dexdor可采用50 mg/ml(5%)葡萄糖注射液、林格氏液、200 mg/ml(20%)甘露醇注射液或9 mg/ml(0.9%)氯化钠注射液稀释至临床给药浓度4mg/ml。已表明Dexdor与以下输液或药物相容:乳酸钠林格氏液、5%葡萄糖注射液、9 mg/ml(0.9%)氯化钠注射液、200 mg/ml(20%)甘露醇、硫喷妥钠、依托咪酯、维库溴铵、潘库溴铵、琥珀酰胆碱、苯磺酸阿曲库铵、咪伐氯铵、罗库溴铵、格隆溴铵、盐酸苯肾上腺素、硫酸阿托品、多巴胺、去甲肾上腺素、多巴酚丁胺、咪达唑仑、硫酸吗啡、枸橼酸芬太尼和血浆代用品。2.2.2.原料药
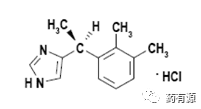
右美托咪定的化学名为1H-咪唑,4-[1-(2,3-二甲基苯基)甲基]-,(S)-,盐酸盐或 (+)-4-[(S)-α,2,3-三甲基苄基]-咪唑盐酸盐,相应分子式为C13H16N2·HCl。原料药结构如图:原料药为白色或类白色结晶粉末,易溶于水、氯仿、乙醇和甲醇,微溶于乙腈,几乎不溶于乙醚。右美托咪定有一个手性中心,因此具有光学活性。原料药为(S)-对映体并被用于制剂生产。已知右旋美托咪定有两种晶型:无水物(A晶型)和一水合物(B晶型)。合成工艺始终获得的为A晶型并用于制剂生产。2.2.2.1. 生产工艺
原料药按活性成份主文件(ASMF)程序申请。右旋美托咪定的制备工艺包括三步合成和纯化(过滤和结晶)。合成路线的完整描述见ASMF的保密部分。对关键步骤和中间体进行了充分地控制以确保原料药质量,并提供了起始物料、试剂和溶剂完善的质量标准。纯化后的原料药装于白色聚乙烯容器并密封保存于铝袋中。制剂制造商质量负责人提供了原料药的生产符合欧盟GMP或ICH Q7A的声明。通过红外光谱、核磁共振氢谱、核磁共振碳谱、紫外光谱和质谱等分析手段确证了原料药的化学结构。此外,通过元素分析确定了分子量,通过X射线衍射确定了绝对构型和晶体结构。2.2.2.2. 质量标准
制剂生产厂对原料药的质量标准包括:性状(目视)、鉴别(IR、氯化物和HPLC)、干燥失重(Ph.Eur.)、硫酸灰分(Ph.Eur.)、重金属(Ph.Eur.)、pH值、溶液的颜色(Ph.Eur.)、溶液的澄清度(Ph.Eur.)、光学纯度(Ph.Eur.)、杂质(HPLC),残留溶剂(GC)、含量(HPLC)和微生物限度(Ph.Eur.)。提供了所有分析方法的详细描述。为内部分析方法提供了完整的方法验证数据,并符合相关ICH指南。总的来说,所提出的分析方法适合于控制活性物质的质量。杂质限值是可以接受的,从安全性的角度来看没有问题。已提供批次分析数据,并显示符合预定义的活性物质规范。2.2.2.3. 稳定性
根据ICH指南,完成了6个生产规模批次的长期(25°C/60%RH)稳定性和5个生产规模批次的加速(40°C/75%RH)稳定性研究,证明原料药具有足够的稳定性。在稳定性研究期间采用放行标准中的分析方法考察了如下指标:性状(目视)、干燥失重、含量(HPLC)、杂质(HPLC)和光学纯度(HPLC)。稳定性研究结果表明原料药储存在原包装中的复验期是合理的。2.2.3.制剂
2.2.3.1. 产品开发
提供资料充分说明了原料药特性和辅料选择合理性。该制剂所选辅料为药物制剂常用辅料并已收入欧洲药典。主要目标是开发一种稳定无菌的供静脉注射的盐酸右美托咪定水溶液。由于原料药在室温下易溶于水,故在目标浓度(100µg/ml)下可将终产品开发为均一的溶液剂。在产品开发过程中考虑并研究了终端灭菌工艺。终产品用镀膜的丁基橡胶塞密封。由于该产品仅供一次性使用,因此未加入防腐剂。所采用的生产工艺简单,且在产品开发过程中没有显著变化。2.2.3.2. 生产
生产过程包括以下步骤:物料混合和溶解、过滤、灌装、终端灭菌、安瓿/小瓶检验、组装(标签和包装)。在产品开发过程中确定并优化制造工艺的关键步骤。提供了生产过程的主要步骤足够的工艺验证数据。批分析结果表明可以该工艺可重复生产出符合要求的成品。2.2.3.3. 质量标准
制剂质量标准是输液用浓缩溶液的标准,对溶液的颜色(Ph.Eur.)、溶液的澄清度(Ph.Eur.)、不溶性微粒(Ph.Eur.)、可抽取体积(Ph.Eur.)、pH(Ph.Eur.)、右美托咪定鉴定(HPLC、UV),含量测定(HPLC)、光学纯度、杂质(HPLC),氯化钠含量、无菌(Ph.Eur.)和细菌内毒素(Ph.Eur.)。从安全的角度对杂质和降解产物进行了评估并认为它们是可接受的。成品检测所用分析方法均进行了恰当的描述,并根据ICH相关指南完成了验证,结合满足要求。5个生产规模批次的批分析数据表明可重复生产出符合要求的输液用浓缩溶液。2.2.3.4. 稳定性
在ICH规定的长期和加速条件下(即25°C/60%RH和40°C/75%RH)对中试和生产批次进行了稳定性研究,涵盖了所有三个申报规格(200μg/2 ml、400μg/4 ml和1000μg/10 ml)。提交以下试验结果:外观(溶液的颜色和澄清度)、杂质、含量、pH、微生物限度纯度(Ph.Eur)以及光学纯度。用于稳定性研究的分析方法与用于成品常规测试的方法相同。稳定性研究期间产品质量没有明显变化。稳定性研究中的所有结果都在质量标准范围内。根据ICH Q1B的建议进行了光稳定性测试。结果符合质量标准要求,成品不需要特殊的光保护。对右美托咪定100μg/ml输液浓缩溶液进行了反复冻融稳定性评价。右美托咪定100μg/ml输液浓缩溶液的冻融研究是将2ml安瓿在大约-20℃的冰箱中至少保存24小时,冻融共进行了四次。所有结果均符合质量标准要求,确认反复冻融对成品的质量和稳定性没有影响。在产品开发过程中,对右美托咪定100μg/ml浓缩液在不同输液中的稳定性和配伍情况进行了研究。研究证明产品经0.9%氯化钠溶液稀释至4μg/ml后48小时稳定,产品经5%葡萄糖(右旋糖)、0.9%氯化钠溶液、林格氏液和20%甘露醇稀释至1和50μg/ml,24小时内稳定。稳定性末期的右美托咪定100μg/ml输液浓缩液经不同稀释液稀释后,在注射器中经室温和2~8℃下保存24小时,所有结果均符合质量标准,表明稀释液不影响成品的质量和稳定性。结果还表明稀释后的溶液在24小时内是稳定的。根据本研究,右美托咪定100μg/ml输液浓缩液与氯化钠、葡萄糖、醋酸林格和甘露醇输液溶液在室温和2-8℃下以4μg/ml的浓度配伍24小时稳定。根据以上稳定性数据,SmPC中规定的有效期和贮藏条件是可以接受的。2.2.4.化学、药剂学和生物学方面的讨论
2.2.4.1. 质量开发
已按照CHMP和ICH相关指导原则,以令人满意的方式呈现了制剂的开发、生产工艺、原料药和制剂的质量控制,并证明了其合理性。生产工艺流程图提供了恰当的过程控制措施。生产工艺在拟定的生产场所对每个规格采用生产规模批次进行了充分的验证。针对成品确定的日常检验用质量标准和测试方法能够充分控制成品的质量。分析方法根据相关指导原则进行了良好的描述和验证。根据提供的稳定性研究数据,容器密封系统能够有效地保障成品的质量。稳定性考察条件符合ICH稳定性指导原则。成品控制检测方法和质量标准充分地建立。 2.2.5.化学、药剂学和生物学方面的结论
有关原料药和制剂的开发、生产和控制的信息已以令人满意的方式提交。所进行的试验结果表明,关键产品质量属性具有令人满意的一致性和均匀性,从而得出结论:该药品在临床上应具有令人满意的一致性和均匀性。在CHMP作出结论时,所有质量方面的问题均已得到解决。
EMA原文链接:
https://www.ema.europa.eu/en/documents/assessment-report/dexdor-epar-public-assessment-report_en.pdf