来源:药学有源
导读
氟维司群(Fulvestrant)是一种雌激素受体拮抗剂。原研药品为阿斯利康开发的氟维司群注射液FASLODEX®(规格50mg/mL,NDA #021344),推荐剂量为500mg/次,在第1、15、29天和之后每月一次肌肉注射给药,单药或联合用药治疗晚期乳腺癌。2019年9月氟维司群注射液被列入国家卫生健康委办公厅发布的我国《第一批鼓励仿制药品目录》(国卫办药政函〔2019〕744号)。
原研药品FASLODEX®为均一溶液,处方中非活性成份包括乙醇、苯甲醇、苯甲酸苄酯和蓖麻油。根据FDA相关技术要求,其仿制药在满足处方种类和浓度与参比制剂相同的条件下,可申请免生物等效性(BE)试验。FDA个药指南给出生物等效性试验建议如下:氟维司群是仅供注射给药的注射剂,根据21 CFR 314.94 (a)(9)(iii)和320.22 (b)(1),如果受试制剂含有与已批准的参比制剂具有相同种类和浓度的活性成份和非活性成份,则可适用豁免体内生物等效性试验。关于21 CFR 320.22 (b)(1)豁免体内生物利用度或生物等效性证据的标准,可参见往期文章FDA审评报告:无醇多西他赛注射液豁免生物等效研究。
那么,如果申报与原研品处方不同的产品呢?FDA基于生物等效性研究,批准了两个505(b)(2) NDA申请,这两个产品与FASLODEX的处方存在差异(NDA#210063/TEVA PHARMS USA INC和NDA#210326/FRESENIUS KABIUSA),具体处方组成如下表。
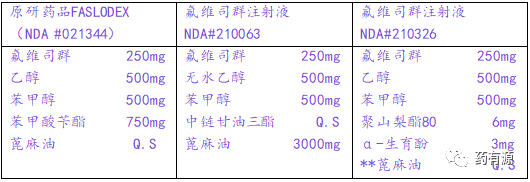
注:**为FDA审评报告资料中隐去内容,下同。
由于氟维司群注射液为肌肉注射的缓释注射液,半衰期约40天,其BE试验存在一定的难度,两个505(b)(2) NDA申请BE研究受试者分别达到252人和266人,研究时间分别长达56天和238天。本文翻译NDA#210326申请的临床药理学审评报告如下:
FDA审评报告原文翻译
摘要
本申请为FreseniusKabi USA,LLC.提交的505(b)(2) NDA申请,申请批准用于肌肉注射50mg/mL的氟维司群注射液,并拟适用于已批准上市的参比制剂FASLODEX®的所有适应症。
申请人提交了在266名健康成年女性受试者中开展的生物等效性研究数据,将申报产品与参比制剂进行对比研究以支持该上市申请。研究结果表明,申报产品与参比制剂具有相似的曲线下面积(AUC)和最大血浆浓度(CMAX)。

1.1 建议
临床药理学办公室已审查了NDA210326,并从临床药理学的角度建议批准50mg/mL氟维司群注射液。
1.2 关键临床药理学研究总结
本申请是根据法案505(b)(2)提交的50mg/mL氟维司群注射液的NDA申请。本申请的参比制剂为FASLODEX®(NDA21344),被批准单药用于治疗未经内分泌治疗的绝经后妇女HR+/HER2-晚期乳腺癌,以及抗雌激素治疗后疾病进展的绝经后妇女HR+晚期乳腺癌,并可与Palbociclib(哌柏西利)或Abemaciclib(玻玛西林)联用治疗内分泌治疗后疾病进展的妇女HR+/HER2-晚期或转移性乳腺癌。
(本申请中的)氟维司群注射液,规格250mg/5mL(50mg/mL),为无色至黄色的溶液,装于一次性无菌预充注射器中,用于肌肉注射。该制剂包括非活性成份:苯甲醇、聚山梨酯80、α-生育酚和**蓖麻油。氟维司群注射剂的处方组成和用量与参比制剂均不同。本申请提交了申报产品相比参比制剂的相对生物利用度数据,以支持批准该产品。
申请人采用正常、健康、不吸烟的绝经后女性为受试者,采用50mg/mL氟维司群注射液与50mg/mL Faslodex®注射液(参比制剂),经肌肉注射,进行了一项开放、双治疗、单周期、平行、单剂量、随机、禁食、实验室盲法的生物等效性研究(研究FULV-006-CP1)。这项研究比较了250mg剂量下氟维司群注射液和250mg剂量下FASLODEX®在注射后长达238天的PK曲线。PK参数(CMAX和AUC)的几何平均比及其90%置信区间在生物等效范围内(见表1)。
研究可靠性和监测办公室(OSIS)建议在不进行现场检查的情况下接受上述数据,因为该场所已接受过检查且检查结果被归类为无需采取行动(见3.1节)。
2 基于问题的审评
2.1 药物的一般特性
2.1.1 原料药和制剂的化学和理化性质的关键点有哪些?
原料药:
氟维司群(C32H47F5O3S)为白色粉末,分子量606.77(图1),几乎不溶于水。
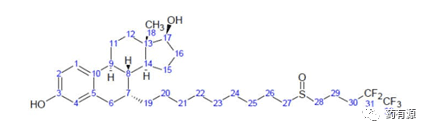
图1:氟维司群的结构
制剂:
氟维司群注射液,250mg/mL,是一种无色至黄色的透明粘性无菌液体,装于一次性无菌预充注射器中,用于肌肉注射,每个注射器含有250mg氟维司群。处方中的非活性成分包括:乙醇、苯甲醇、聚山梨酯80、α-生育酚和**蓖麻油。
2.1.2 申请的作用机制和治疗适应症是什么?
氟维司群是一种抗雌激素的药物,以竞争的方式与雌激素受体(ER)结合。体外试验显示氟维司群可抑制雌激素敏感和他莫昔芬耐药的人乳腺癌细胞的生长。
氟维司群注射剂适用于未经内分泌治疗的绝经后妇女HR+/HER2晚期乳腺癌的治疗,以及内分泌治疗后病情进展的绝经后妇女HR+/**晚期乳腺癌的治疗。氟维司群还可联用Palbociclib(哌柏西利)用于治疗内分泌治疗后疾病进展的妇女HR+/HER2晚期乳腺癌。
2.1.3 申请的剂型和给药途径是什么?
氟维司群注射液包括两针5ml的预充注射器,每个注射器含有250mg氟维司群。推荐剂量为500mg,在第1、15、29天和之后每月一次经肌肉注射给药,每次注射两针5ml,缓慢地(每次注射1-2分钟)至臀部(臀区),两侧臀部各注射一针。
2.2 一般临床药理学
2.2.1 用于支持给药或声明的临床药理学和临床研究的设计特点是什么?
为了支持该505(b)(2)NDA申请,申请人进行了一项关键的生物等效性研究和一项试验性相对生物利用度研究。表2总结了这两项研究。

申请人进行了一项开放标签、随机、双治疗、平行、单剂量生物等效性研究(FULV-005-CP1)的试验性研究,以考察申报产品相对参比制剂的相对生物利用度。研究的目的是确定申报产品PK参数的变异性,以确定关键研究的足够样本量,评估注射部位的疼痛,并监测研究对象的安全性。
关键的BE研究(FULV-006-CP1)是一项针对50mg/mL氟维司群注射液的开放标签、随机、平行、单剂量的生物等效性研究,采用健康、成人、不吸烟女性受试者在禁食条件下试验。次要目标是评估注射部位的疼痛并监测研究对象的安全性。
2.2.2 药物的PK特征是什么?
以下信息来自参比制剂FASLODEX®当前批准的标签。
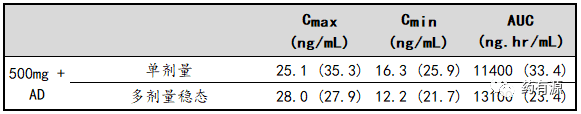
吸收——表3总结了500mg给药方案的单剂量和多剂量PK参数,第15天增加一次给药(AD)以达到稳态浓度。
表3:绝经后晚期乳腺癌患者肌肉注射500mg+AD方案后氟维司群-PK参数[gMean(CV%)]
分布——稳态时的表观分布体积约为3至5L/kg。主要分布在血管外。氟维司群与血浆蛋白高度结合(99%),VLDL、LDL和HDL脂蛋白组分表现为主要的结合成分。
代谢——氟维司群生物转化涉及多种途径:氧化、芳香羟基化、与葡萄糖醛酸和/或硫酸盐结合。在抗雌激素模型中,所鉴定的代谢物活性较低或与氟维司群表现出相似的活性。细胞色素P4503A4(CYP 3A4)是唯一参与氟维司群氧化代谢的P-450同工酶。
排泄——氟维司群通过肝胆途径快速清除,通过粪便排泄(~90%)。肾清除可忽略不计(小于1%)。肌肉注射250mg后,清除率(平均值±SD)为690±226mL/min,表观半衰期为40天。
2.3 一般生物药剂学
2.3.1 申报产品与已批准的参比制剂的相对生物利用度是多少?
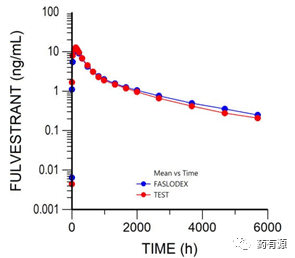
按计划进行的238天的PK取样结果显示,在250mg剂量下氟维司群注射液的AUC和CMAX与FASLODEX®等效。
FULV-006-CP1是一项开放、随机、平行、单剂量的生物等效性研究,以健康、成人、不吸烟女性为受试者,在禁食条件下注射50mg/mL的氟维司群注射液。PK取样在给药后进行长达238天。
研究显示申报产品与参比制剂具有生物等效性(图2和表1)。所提出的公式的PK申报产品的PK参数的几何平均比以及90%的置信区间均在80%到125%之间。
2.4 分析部分
2.4.1 如何在血浆中鉴别和测量活性成份?
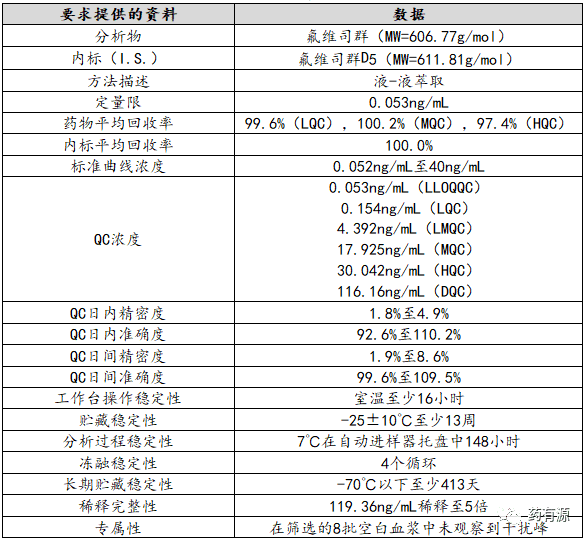
采用高效液相色谱-质谱联用技术测定血浆中的氟维司群。样品制备采用液-液萃取技术。
2.4.2 对于所有检测部分,游离、结合或总量是否均检测?
氟维司群总浓度在关键BE试验(FULV-006- CP1)中测定。
2.4.3 使用什么生物分析方法来测定浓度?
采用高效液相色谱-串联质谱法(HPLC-MS/MS)测定了氟维司群的浓度。
2.4.4 标准曲线的范围是多少?与临床试验要求的相关性如何?采用什么曲线拟合方法?
标准曲线的浓度范围为0.052 ng/mL至40ng/mL。浓度范围涵盖了观测到的平均CMAX(~13ng/mL)和PK最后一个取样时间点的平均浓度(第238天为0.2ng/mL)。采用加权(1/x2)线性回归法对氟维司群浓度进行反算。
2.4.4.1 定量上下限是多少?
LLOQ为0.054 ng/mL,ULOQ为40 ng/mL。
2.4.4.2 这些限值的准确度、精密度和选择性是多少?
准确度范围为90.6%至100.8%,在LLOQ和ULOQ的精密度范围为1.8%至5.5%。
2.4.4.3 试验条件下样品稳定性如何?
在室温、冻融循环(4个循环)、使用条件(至少148小时)、短期稳定性(25℃至少6小时)和长期稳定性(-70℃至少417天)条件下评估QC样品的稳定性。
2.4.4.4 QC取样计划是怎么样的?
每次试验均包括在LLOQ(0.054ng/mL)、低浓度(~0.15 ng/mL)、低至中浓度(~4.5ng/mL)、中浓度(~18ng/mL)和高浓度(~30ng/mL)的QC样品。试验内和试验间的准确度和精密度值均符合法规可接受标准(表4)。
FDA原文链接:
相关文章
