
伏硫西汀(Vortioxetine)由灵北和武田制药联合研发,2013年9月30日获FDA批准上市,适应症为抑郁症。2013年12月18日获欧盟EMA批准上市,剂型为口服薄膜包衣片和滴剂,商品名“TRINTELLIX”。
2.2 Quality aspects 质量部分
The finished product is presented as immediate-release film-coated tablets containing 5, 10, 15 or 20mg of Vortioxetine (as hydrobromide salt) as the active substance and as oral drops containing 20mg/ml of Vortioxetine (as the lactate salt) as the active substance.
该产品包括含有5、10、15或20mg伏硫西汀(氢溴酸盐)的速释薄膜包衣片,以及含有20mg伏硫西汀(乳酸盐)的口服滴剂。
Other ingredients in the film-coated tablets are mannitol (E421), microcystalline cellulose, hydroxypropylcellulose, sodium starch glycolate (type A) and magnesium stearate which are present in the tablet core and hypromellose, macrogol 400, titanium dioxide (E171) and iron oxide red (E172) and/or iron oxide yellow (E172) present in the tablet coating.
薄膜包衣片的片芯中含有的其他成分包括甘露醇(E421)、微晶纤维素、羟丙纤维素、羧甲基淀粉(A型)和硬脂酸镁,包衣成分包括羟丙甲纤维素、聚乙二醇400、二氧化钛或氧化铁黄(E172)。
Other ingredients in oral drops are hydroxypropylbetadex, ethanol (96 %) and purified water.口服滴剂所含有其他成分包括羟丙基-β-环糊精、乙醇(96%)和纯化水。
The tablets are available in PVC/PVdC blisters or white HDPE bottles with child-proof closure and tamper-evident seal. The oral drops are packed in amber glass bottles with dropper applicator (low density polyethylene), and child-proof screw cap (polypropylene) as described in section 6.5 of the SmPC.
片剂采用PVC/PVdC泡罩包装,或采用具有防儿童开启和防拆封功能的HDPE瓶包装。口服滴剂采用产品说明书6.5所述的具有滴管(低密度聚乙烯)的安瓿玻璃瓶和具有防儿童开启螺旋盖(聚乙烯)包装。
Vortioxetine has been qualified as a new active substance. Its salts, Vortioxetine hydrobromide and Vortioxetine DL-lactate have been used to manufacture the immediate-release film-coated tablets and the oral drops respectively.
伏硫西汀(Vortioxetine)已证实为新活性成分,采用伏硫西汀氢溴酸盐和伏硫西汀DL-乳酸盐分别制备了速释薄膜包衣片和口服滴剂。
Vortioxetine hydrobromide
氢溴酸伏硫西汀
The chemical name of Vortioxetine hydrobromide is 1-[2-(2,4-dimethyl-phenylsulfanyl)-phenyl]- piperazine hydrobromide and has the following structure:
氢溴酸伏硫西汀的化学名称为1-[2-(2,4-二甲基-苯基磺胺基)-苯基]-哌嗪氢溴酸盐,其结构如下:
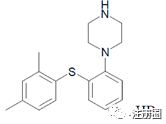
The molecular formula is C18H22N2S.HBr and its relative molecular mass is 298.45 mg/mol as a free base and 379.36 mg/mol as the hydrobromide salt. Vortioxetine hydrobromide appears as a white to very slightly beige powder, non-hygroscopic, soluble in methanol and ethanol and slightly soluble in water and aqueous solutions at pH 2.0 to 8.3. Its pKa is
9.1 as the free base and 3.0 as the salt. Vortioxetine exhibits polymorphism and appears in four polymorphs. The most thermodynamically stable polymorphic form has been determined and the crystallisation process is designed to consistently deliver this form. Vortioxetine has a non-chiral molecular structure.
分子式为C18H22N2S·HBr,游离碱摩尔质量为298.45mg/mol,氢溴酸盐为379.36mg/mol。氢溴酸伏硫西汀为白色至极浅褐色粉末,无吸湿性,溶于甲醇和乙醇,微溶于水,水溶液pH值为2.0至8.3。游离碱pKa为9.1、盐为3.0。伏硫西汀具有4种多晶型。已经确定了热力学上最稳定的晶型,并且设计了结晶过程以确保始终获得这种晶型。伏硫西汀不具有手性中心。
Manufacture 生产
Vortioxetine hydrobromide is manufactured in two well defined synthetic steps, followed by recrystallization and milling. The starting materials used are well defined and commercially available with acceptable specifications. Two sites are involved in the manufacture of Vortioxetine hydrobromide. The route of synthesis has been described in sufficient detail and adequate in-process controls are applied during the synthesis. The specifications and control methods for intermediate products, starting materials and reagents have been presented. Information about the formation, presence, origin and fate of impurities during manufacture has been satisfactorily discussed. The characterisation of the active substance and its impurities are in accordance with the EU guideline on chemistry of new active substances. Potential and actual impurities were well discussed with regards to their origin and characterised. The specifications and control methods for intermediate products, starting materials and reagents have been presented.
伏硫西汀氢溴酸盐通过两个明确定义的合成步骤生产,然后进行重结晶和研磨。所使用的起始原料定义明确,并且从可接受的合格供应商获得。伏硫西汀氢溴酸盐的生产涉及两个生产场所。已经充分详细地描述了合成工艺并进行了充分的过程控制。明确了中间产品、起始原料和试剂的质量标准和控制方法。充分地讨论了生产过程中杂质的形成、存在、来源和清除过程。活性物质及其杂质的表征符合欧盟关于新活性物质的指南。关于潜在杂质和实际存在杂质的来源和特性进行了充分讨论。
Representative batch analysis data provided for the two proposed manufacturing sites produced with the proposed synthetic route show that the active substance can be manufactured reproducibly.
提供的两个拟定的生产场所的代表性批次分析数据显示,采用拟定的合成工艺可重复生产出符合要求的活性物质。
Specification 质量标准
The active substance specification includes tests for identification (HPLC, FTIR and NIR), assay (HPLC), impurities (HPLC and GC), residual solvents (GC), residue on ignition, heavy metals and particle size. The analytical methods used have been adequately described and non-compendial methods appropriately validated in accordance with the ICH guidelines.
原料药的质量标准包括鉴别(HPLC、FTIR和NIR)、含量测定(HPLC)、有关物质(HPLC和GC)、残留溶剂(GC)、炽灼残渣、重金属和粒径。详细描述了分析方法,非药典分析方法已根据ICH指南进行了充分验证。
Batch analysis data for a number of commercial and development batches of the active substance are provided. The results are within the specifications and consistent from batch to batch.
提供了多批商业批和开发批次原料药的批分析数据,均符合质量标准且批间一致性良好。
Stability 稳定性
Stability data were provided on 2 pilot and 9 production scale batches of Vortioxetine hydrobromide active substance from the proposed manufacturers stored in a container closure system representative of that intended for the market. According to the ICH guidelines the samples were stored up to 48 months under long term conditions at 25ºC/60% RH and for up to 6 months under accelerated conditions at 40ºC/75% RH. Photostability testing following the ICH guideline Q1B was performed on one batch. Results on stress conditions 60°C/80% RH were also provided on one batch.
提供了按申报工艺生产并采用拟上市包装的2批中试批和9批商业批氢溴酸伏硫西汀原料药的稳定性数据。根据ICH指南提供了上述批次长期(25℃/60%RH)48个月和加速(40℃/75%RH)6个月的稳定性数据。采用1批原料药根据ICH Q1B进行了光稳定性研究,并采用1批原料药在60℃/80%RH条件下进行了强制降解试验。
The following parameters were tested: description, assay and impurities. The analytical methods used were the same as for release and were stability indicating. The stability results indicate that the active substance manufactured by the proposed suppliers is sufficiently stable. The stability results justify the proposed retest period in the proposed container
对以下属性进行了考察:性状、含量和有关物质。所采用的分析方法同放行标准且可以反应稳定性的变化。稳定性数据表明采用申报工艺生产的原料药稳定,且证明拟定包装中的复验期是合理的。
Vortioxetine DL-lactate
伏硫西汀DL-乳酸盐
Vortioxetine DL-lactate is a lactate salt of vortioxetine and its chemical name is 1-[2-(2,4-dimethyl phenylsulfanyl)-phenyl]-piperazine (RS)-2-hydroxypropanoate and has the following structure:
伏硫西汀DL-乳酸盐化学名称为1-[2-(2,4-二甲基-苯基硫烷基)-苯基]-哌嗪(RS)-2-羟基丙酸酯,并具有以下结构:
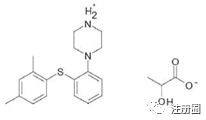
The molecular formula is C18H22N2S.C3H6O3 and its relative molecular mass as the lactate salt is 388.52 mg/mol. The lactate salt of vortioxetine is used for the oral drops formulation due to its increased solubility in polar solvents.
分子式为C18H22N2S.C3H6O3,相对摩尔质量为388.52mg/mol。由于乳酸盐在极性溶剂中较好的溶解度而用于滴剂。
Vortioxetine lactate appears as a white to beige powder, non-hygroscopic, soluble in polar solvents and water. Its pKa is 9.1 as the free base and 3.0 as the salt.
Vortioxetine exhibits polymorphism and appears in three polymorphs. The most thermodynamically stable polymorphic form has been determined and the crystallisation process is designed to consistently deliver this form.
伏硫西汀乳酸盐为白色至米色粉末,无吸湿性,溶于极性溶剂和水,游离碱pKa为9.1、盐为3.0。伏硫西汀具有3种多晶型。已经确定了热力学上最稳定的晶型,并且设计了结晶过程以确保始终获得这种晶型。
Manufacture
生产
Vortioxetine DL-lactate is manufactured from the milled Vortioxetine hydrobromide via the non isolated free base. One site is involved in the manufacture of the lactate salt of Vortioxetine.
伏硫西汀DL-乳酸盐是由经研磨的氢溴酸伏硫西汀通过非分离的游离碱制得的。伏硫西汀乳酸盐的生产在一个生产场所进行。
The route of synthesis has been described in sufficient detail and adequate in-process controls are applied during the synthesis. The specifications and control methods for intermediate products, starting materials and reagents have been presented. Information about the formation, presence, origin and fate of impurities during manufacture has been satisfactorily discussed.
充分详细地描述了合成过程及过程控制措施。提出了中间产品、起始原料和试剂的质量标准和控制方法。充分地讨论了生产过程中杂质的形成、存在、来源和清除过程。
The characterisation of the active substance and its impurities are in accordance with the EU guideline on chemistry of new active substances. Potential and actual impurities were well discussed with regards to their origin and characterised.
活性物质及其杂质的表征符合欧盟关于新活性物质的指南。关于潜在杂质和实际存在杂质的来源和特性进行了充分讨论。
Representative batch analysis data provided for the proposed manufacturing site produced with the proposed synthetic route show that the active substance can be manufactured reproducibly
提供的拟定的生产场所的代表性批次分析数据显示,采用拟定的合成工艺可重复生产出符合要求的活性物质。
Specification
质量标准
The active substance specification includes tests for identification (HPLC, FTIR), assay (HPLC), impurities (HPLC and GC), residual solvents (GC), residue on ignition, heavy metals and particle size.
原料药的质量标准包括鉴别(HPLC、FTIR)、含量测定(HPLC)、有关物质(HPLC和GC)、残留溶剂(GC)、灼烧残渣、重金属和粒径。
The analytical methods used have been adequately described and non-compendial methods appropriately validated in accordance with the ICH guidelines.
详细描述了分析方法,非药典分析方法已根据ICH指南进行了充分验证。
Batch analysis data for four commercial and development batches of the active substance are provided. The results are within the specifications and consistent from batch to batch.
提供了多批商业批和开发批次原料药的批分析数据,均符合质量标准且批间一致性良好。
Stability
稳定性
Stability data were provided on three pilot batches of Vortioxetine DL-lactate active substance from the proposed manufacturer stored in the intended commercial package. According to the ICH guidelines the samples were stored up to 48 months under long term conditions at 25ºC/60% RH and for up to 6 months under accelerated conditions at 40ºC/75% RH.
提供了按申报工艺生产并采用拟上市包装的3批中试批伏硫西汀乳酸盐原料药的稳定性数据。根据ICH指南提供了上述批次长期(25℃/60%RH)48个月和加速(40℃/75%RH)6个月的稳定性数据。
Photostability testing following the ICH guideline Q1B was performed on one batch. Results on stress conditions 60°C/80% RH were also provided on one batch.
采用1批原料药根据ICH Q1B进行了光稳定性研究,并采用1批原料药在60℃/80%RH条件下进行了强制降解试验。
The following parameters were tested: description, assay and impurities. The analytical methods used were the same as for release and were stability indicating.
The stability results indicate that the active substance manufactured by the proposed suppliers is sufficiently stable. The stability results justify the proposed retest period in the proposed container.
对以下属性进行了考察:性状、含量和有关物质。所采用的分析方法同放行标准且可以反应稳定性的变化。稳定性数据表明采用申报工艺生产的原料药稳定,且证明拟定包装中的复验期是合理的。
Film-coated tablets
薄膜包衣片
Pharmaceutical Development
制剂开发
Vortioxetine immediate-release film-coated tablets contain 5, 10, 15 or 20 mg of the Vortioxetine (as hydrobromide salt).
伏硫西汀速释薄膜包衣片包含5、10、15或20mg伏硫西汀(氢溴酸盐)。
During the pharmaceutical development the applicant evaluated different formulations to find the most appropriate combination of excipients and physicochemical and biological properties of the formulation/blend to manufacture the tablet with the best possible tablet characteristics and dissolution rate. The dissolution rate is influenced by the particle size and therefore the active substance is milled to obtain the required particle size distribution throughout the blend.
制剂开发过程中,申请人对不同处方进行了评估以筛选出最适宜的辅料组成,以及理化和生物学性质、片剂属性和溶出度最佳的处方。制剂的溶出度受粒径影响,因此对原料药进行了研磨以在混合工艺中获得适宜的粒度分布。
The compatibility of the active substance with the excipients has been evaluated using binary mixtures in stability studies. All excipients are well known pharmaceutical ingredients and their quality is compliant with Ph. Eur. standards. There are no novel excipients used in the finished product formulation. The list of excipients is included in section 6.1 of the SmPC.
通过二元混合试验证明原辅料相容性良好。所有辅料均为药用成分并符合欧洲药典要求。制剂中未使用新辅料。所用辅料清单列在说明书6.1部分。
The formulation used during clinical studies is the same that the used for marketing. The development of the manufacturing process of film-coated tablets 5 mg, 10 mg, 15 mg and 20 mg was performed at pilot scale on similar equipment used for production scale.
临床试验中使用的处方与申报上市处方相同。5mg、10mg、15mg和20mg薄膜包衣片工艺开发的中试批样品采用与生产规模相似的设备生产。
The film-coated tablets are packed in polyvinyl chloride/polyvinylidene chloride/aluminium (PVC/PVdC/alu) blisters and in high density polyethylene (HDPE) containers. The material complies with Ph. Eur. and EC requirements. The choice of the container closure system has been validated by stability data and is adequate for the intended use of the product.
薄膜包衣片采用PVC/PVdC铝塑泡罩和HDPE包装。包装材料符合欧洲药典和EC要求。通过稳定性研究说明了容器包装系统的选择依据,并与产品预期用途相符。
Adventitious agents
外来物质
No excipients derived from animal or human origin have been used.
未使用动物源或人源辅料。
Manufacture of the product
制剂生产
The manufacturing process of the immediate release dosage form involves the following steps: blending, fluid bed granulation, drying, blending, compression and film coating.
The process is considered to be a standard manufacturing process.
该速释制剂的生产过程包括以下步骤:混合、流化床制粒、干燥、混合、压片和包衣。该生产过程可认为是标准生产过程。
Major steps of the manufacturing process have been validated by a number of studies. It has been demonstrated that the manufacturing process is capable of producing the finished product of intended quality in a reproducible manner. The in-process controls are adequate for this type of manufacturing process.
主要生产步骤经过一系列研究进行了验证。已证明生产过程可持续生产出符合质量要求的制剂。针对该生产工艺进行了充分的生产过程控制。
Product specification
制剂质量标准
The finished product release specifications include appropriate tests for this kind of dosage form: description (visual), identification (HPLC, UV and FT-IR), uniformity of dosage units, assay (HPLC), degradation products (HPLC), dissolution, water content and microbiological quality (Ph. Eur.).
制剂放行标准包括此类剂型的检查项,包括性状(目视)、鉴别(HPLC、UV和FT-IR)、含量均匀度、含量(HPLC)、降解产物(HPLC)、溶出度、水分和微生物限度(Ph. Eur.)。
The analytical methods used have been adequately described and non-compendial methods appropriately validated in accordance with the ICH guidelines.
Batch analysis results are provided for three pilot scale batches for each strength, confirming the consistency of the manufacturing process and its ability to manufacture to the intended product specification.
详细描述了分析方法,非药典分析方法已根据ICH指南进行了充分验证。提供了每个规格3个中试批产品的批分析结果,证明了生产工艺的一致性和可生产出符合预期质量标准产品的稳定性。
Stability of the product
制剂稳定性
Stability data of three pilot scale batches of finished product stored under long term conditions for 24 months at 25ºC/60% RH and intermediate conditions at 30°C/75% RH, and for up to 6 months under accelerated conditions at 40ºC/75% RH according to the ICH guidelines were provided. The batches of medicinal product are representative to those proposed for marketing and were packed in the primary packaging proposed for marketing.
根据ICH指南,提供了3批中试规模制剂长期条件(25℃/60%RH)和中间条件(30℃/75%RH)24个月,以及加速条件(40℃/75%RH)6个月的稳定性数据。进行稳定性研究的制剂批次可以代表拟上市产品并采用拟上市初级包装。
Samples were tested for tablet description, assay, degradation products, dissolution, water content and microbiological quality. The analytical procedures used are stability indicating.
考察了片剂的性状、含量、降解产物、溶出度、水分和微生物限度。所采用的分析方法可以反应稳定性的变化。
In addition, one batch was exposed to light as defined in the ICH Guideline on Photostability Testing of New Drug Substances and Products and also to a humidity stress test during 3 months at 25°C/93% RH.
此外,采用1批制剂根据ICH新原料药和制剂光稳定性指南进行的光暴露研究,并在25℃/93%RH条件下进行了3个月的高湿考察。
Based on available stability data, the shelf-life and storage conditions as stated in the SmPC are acceptable.
根据提供的稳定性数据,产品使用说明书中规定的有效期和贮藏条件是可接受的。
Oral drops
口服滴剂
Pharmaceutical development
制剂开发
The aim was to develop an oral drops formulation with a concentration of 20 mg vortioxetine per ml to be administered with a dropper device corresponding to 20 drops for a dose of 20 mg vortioxetine.
预期目标为开发20mg/ml的口服滴剂且采用滴管给药20滴时剂量为20mg伏硫西汀。
Unlike the film-coated tablets where a hydrobromide salt of the active substance is used, the oral drops formulation contains vortioxetine DL-lactate as the active substance which shows higher solubility in polar solvents. The solubility is further increased due to the presence of hydroxypropylbetadex in the formulation. The substitution degree and the temperature necessary for interaction with the active substance were investigated and taken into consideration during the development. Both parameters were not found critical for the interaction between hydroxypropylbetadex and vortioxetine.
与薄膜包衣片采用氢溴酸盐作为活性成分不同,口服滴剂采用DL-乳酸盐作为活性成分,该活性成分在极性溶剂中具有更高的溶解度。制剂处方中的羟丙基-β-环糊精进一步提高了原料药的溶解度,在开发过程中考察了该辅料的取代度及其与活性成分相互作用所需温度,两个参数对羟丙基-β-环糊精和伏硫西汀的相互作用均不关键。
The other excipient, ethanol 96%, increases the number of drops per ml and at the same time serves as preservative. Various ratios were investigated and a concentration of 8.5% (w/v) gave the desired drop number of 20 drops/ml. The oral drops formulation is self-preserving which is confirmed with a test for efficacy of antimicrobial preservation. Purified water serves as vehicle for the formulation.
另一辅料96%乙醇增加了每毫升溶液的滴数并同时作为防腐剂。在8.5%(w/v)浓度下考察了不同比例用量并获得预期的20滴/毫升。通过抑菌效力试验证明口服滴剂的处方自身具有防腐作用。纯化水为该处方的溶剂。
All excipients are well known pharmaceutical ingredients and their quality is compliant with Ph. Eur. standards. There are no novel excipients used in the finished product formulation. The list of excipients is included in section 6.1 of the SmPC.
所有辅料均为药用成分并符合欧洲药典要求。制剂中未使用新辅料。所用辅料清单列在说明书6.1部分。
The formulation used in the clinical bioequivalence study was the same as the formulation used for marketing. The study also showed bioequivalence between the oral drops solution, 20 mg/ml and the 20 mg film-coated tablet formulation.
临床生物等效性试验所用的处方与申报上市处方相同,其结果证明20mg/ml口服滴剂溶液与20mg规格薄膜包衣片等效。
The primary packaging is an amber glass bottle with dropper applicator and child-proof screw cap. The material complies with Ph. Eur. and EC requirements. The choice of the container closure system has been validated by stability data and is adequate for the intended use of the product.
口服滴剂初级包装为安瓿玻璃瓶和具有防儿童开启功能的螺旋盖。包装材料符合欧洲药典和EC要求。通过稳定性研究说明了容器包装系统的选择依据,并与产品预期用途相符。
Adventitious agents
外来物质
No excipients derived from animal or human origin have been used.
未使用动物源或人源辅料。
Manufacture of the product
制剂生产
The manufacturing process consists of three main steps: preparation of the bulk solution, filtration and subsequent filling of the amber bottles. The manufacturing process is carried out protected from light.
生产工艺包括三个主要步骤:配液、过滤和安瓿瓶灌装。生产过程需要避光。
The process is considered to be a standard manufacturing process. Major steps of the manufacturing process have been validated by a number of studies. It has been demonstrated that the manufacturing process is capable of producing the finished product of intended quality in a reproducible manner. The in-process controls are adequate for this type of manufacturing process.
该生产过程可认为是标准生产过程。主要生产步骤经过一系列研究进行了验证。已证明生产过程可持续生产出符合质量要求制剂。针对该生产工艺进行了充分的生产过程控制。
Product specification
制剂质量标准
The finished product release specifications include appropriate tests for this kind of dosage form and include description (visual), identification (HPLC, UV), assay vortioxetine (HPLC), assay ethanol 96% (GC), degradation products (HPLC), dose and uniformity of dose of oral drops, pH (Ph. Eur.) and microbial quality( Ph. Eur.).
制剂放行标准包括此类剂型的检查项,包括性状(目视)、鉴别(HPLC、UV)、伏硫西汀含量(HPLC)、96%乙醇(GC)、降解产物(HPLC)、口服滴剂的剂量和剂量均一性、pH(Ph. Eur.)和微生物限度(Ph. Eur.)。
The analytical methods used have been adequately described and non-compendial methods appropriately validated in accordance with the ICH guidelines.Batch analysis results are provided for three batches confirming the consistency of the manufacturing process and its ability to manufacture to the intended product specification.
详细描述了分析方法,非药典分析方法已根据ICH指南进行了充分验证。提供了3个批次产品的批分析结果,证明了生产工艺的一致性和可生产出符合预期质量标准产品的稳定性。
Stability of the product
制剂稳定性
Stability data of two production scale and one pilot scale batches of finished product stored under long term conditions for 18 months at 25ºC/60% RH and intermediate conditions at 30°C/75% RH, and for up to 6 months under accelerated conditions at 40ºC/75% RH according to the ICH guidelines were provided. The batches of medicinal product are representative to those proposed for marketing and were packed in the primary packaging proposed for marketing.
根据ICH指南,提供了2批商业生产规模和1批中试规模制剂长期条件(25℃/60%RH)和中间条件(30℃/75%RH)18个月,以及加速条件(40℃/75%RH)6个月的稳定性数据。进行稳定性研究的制剂批次可以代表拟上市产品并采用拟上市初级包装。
Samples were tested for description, vortioxetine assay, ethanol assay, degradation products, dose and uniformity of dose of oral drops, pH, colour of solution and microbiological quality. The analytical procedures used are stability indicating.
考察了制剂的性状、伏硫西汀含量、乙醇含量、降解产物、口服滴剂的剂量和剂量均一性、pH、溶液颜色和微生物限度。所采用的分析方法可以反应稳定性的变化。
In addition, one batch was exposed to light as defined in the ICH Guideline on Photostability Testing of New Drug Substances and Products and in–use stability studies were performed on two batches stored for up to 8 weeks after first opening of the bottles at 25°C/60% RH.
Based on available stability data, the shelf-life and storage conditions as stated in the SmPC are acceptable.
此外,采用1批制剂根据ICH新原料药和制剂光稳定性指南进行的光暴露研究,并采用2批制剂在药瓶首次开启后在25℃/60%RH条件下进行了长达8周使用中稳定性研究。根据提供的稳定性数据,产品使用说明书中规定的有效期和贮藏条件是可接受的。
Information on development, manufacture and control of the active substance and finished product has been presented in a satisfactory manner. The results of tests carried out indicate consistency and uniformity of important product quality characteristics, and these in turn lead to the conclusion that the product should have a satisfactory and uniform performance in clinical use.
关于原料药和制剂的开发、生产和控制的资料已经以令人满意的方式提交。提供的试验结果表明产品关键质量属性的一致性和均匀性,进而证明该产品在临床应用中具有令人满意且均一的性能。
The quality of this product is considered to be acceptable when used in accordance with the conditions defined in the SmPC. Physicochemical and biological aspects relevant to the uniform clinical performance of the product have been investigated and are controlled in a satisfactory way
当按照产品使用说明书中规定的条件使用时,本产品的质量被认为是可接受的。已对与产品临床性能相关的物理化学和生物学方面进行了研究,并以令人满意的方式进行了控制。
© 版权声明
文章版权归作者所有,未经允许请勿转载。
相关文章



暂无评论...