文章来源:研如玉
编者语:我们公众号发表过多篇对具体品种单独规格生物等效性豁免的讨论文章的翻译稿。本篇综述丰富了速释和控释口服固体剂型不同规格的生物等效性豁免中关于EMA与FDA的一些要求和看法。目的是研究EMA对其他规格生物豁免的各种要求,重点是仿制药产品上市的申请。该综述还将比较EMA生物豁免要求和FDA的方法。我们可以看到欧盟和美国监管机构对此的不同要求。提示我们需要区别对待。以符合申报要求。
原文出处见下面截图:

请用手机打开本文,拉到文章最后出现广告页面(如下图)点击下广告。支持下本公众号的付出。谢谢!!!
仿制药实施其他规格生物等效性豁免:EMA建议方法以及美国FDA提交的挑战
J.-M. Cardota,*, A.Garcia-Arietab,1, P. Paixaoc,1, I. Tasevskad,1,B. Davite
aFaculté de Pharmacie, Université ClermontAuvergne, Laboratoire de Biopharmacie, MEDIS, 28 Place H. Dunant, 63001Clermont-Ferrand, France
bPharmacokinetics and Generic Medicines,Division of Pharmacology and Clinical Evaluation, Department of Human UseMedicines, Agencia Española de Medicamentos y Productos Sanitarios (AEMPS),Calle Campezo 1—Edificio 8, 28022 Madrid, Spain
cINFARMED—National Authority of Medicinesand Health Products, Av. do Brasil 53, 1749-004 Lisbon, Portugal
dState Institute for Drug Control (SÚKL),Šrobárova 48, 100 41 Praha 10, Czech Republic
eTranslational Medicine, Merck & Co.,2000 Galloping Hill Road, Kenilworth, NJ 07033, USA
文章信息 | 摘要 |
关键词: 规格 生物豁免 EMA US-FDA | 这篇综述描述了速释和调释口服固体剂型针对其他规格仿制药申请中EMA关于生物豁免的要求,并强调同时提交EMA和FDA的挑战。对目前EMA指南与目前FDA工业指南的一些具体内容进行比较,特别关注体内研究的规格、生物豁免的配方适应性,以及优化其他规格生物豁免的溶出度研究。在欧洲,适用于仿制药的相同原则可以考虑用于推导创新产品的生物豁免。介绍了几个案例研究来说明在EMA和FDA同时申请其他规格生物豁免的挑战。 |
1.介绍
优化药物疗法的能力通常取决于药物产品线多种规格的可用性,以便根据患者的个体需求来给药。个体患者吸收、分布、代谢和清除给定药物的方式取决于内在因素(如年龄、性别、种族、体重、体表面积、代谢状态、肾脏和肝脏损害),以及外在因素(如伴随药物、病理学、与食物共同施用以及药物副作用)。例如,对于主要由肾脏消除的药物,多种规格的可用性可提供肾功能不全病例中降低剂量的能力。 对于其他药物,可能有必要调整剂量,以达到疗效和安全性之间的最佳平衡。 多种规格的可用性对于需要滴定的药物至关重要; 即开始使用低剂量,逐渐递增至较高剂量,以避免急性毒性。 在某些情况下,其他规格的开发可提高依从性,并避免了用药错误的风险。 例如,可以开发较低的规格以避免掰片(特别对于老年人,这是一项复杂的任务),并且可能会影响剂量的均匀性。应该指出,开发多种规格的一个特殊挑战是固定剂量组合,其中两种或更多种药物以相同剂型组合。这样的固定剂量组合可以开发为单一单位片剂、分层片剂或丸剂的组合;这些配方类型中的每一种都提出了自己的一套独特挑战,以有效地配制与剂量成比例的不同规格。
在开发一种新药物产品时,监管者应尽可能避免不必要的人体测试。在这个范围内,欧洲药品管理局(EMA)和美国食品药品监督管理局(FDA)等不同司法管辖区的指导方针提到了豁免体内研究要求的机制——即生物豁免——其他规格(EMA,2010;FDA,2013a,2013b,2014,2015a)。这种其他规格的生物豁免必须保证不同规格具有相同的生物利用率(速率和程度),因此,为了实施这些生物豁免,必须满足许多要求。
制药公司通常同时向多个监管机构提交相同的档案,以最终在多个辖区进行销售。只要有可能则尽量避免重复进行体内研究。尽管国际协调努力,但仍有一个主要障碍,即需要使用当地对照药物或参比药物进行临床相对生物利用度或生物等效性(BA/BE)研究。根据1984年针对食品药品和化妆品法案的Hatch-Waxman修正案,FDA需要每月发布和更新参比药物列表(RLD),列出新仿制药物需要与之进行比较以显示其具有生物等效性的经过批准的药物产品(美国FDA,2017a)。EMA不公布这样的列表,则可能会考虑几种不同的对照药品。这包括使用国家授权程序批准的药物产品,甚至导致各国之间对照组分上可能存在差异。因此,需要针对多个对照药品(FDA RLD和欧盟EU中可能的对照药品之一)进行多项生物利用度/生物等效性研究,这反过来会增加产品开发成本,并可能导致产品在市场上实质性延期(Gwaza等,2016)。第二个障碍源于以下事实:不同的管辖区有不同的地方监管要求,其决定了在体内生物等效性研究中没有进行测试的其他规格的生物豁免的获得。因此,可能有必要开发药品生产线各种规格的不同配方,以满足各管辖区内豁免生物等效性要求的差异。因此,设计符合EMA和FDA监管机构不同要求的药品开发战略对寻求全球市场的药品开发商来说是一项重大挑战。
基于BCS的生物豁免未包含在本文的范围内,因为当基于BCS生物豁免申请的产品存在一系列规格时,EMA Q&A(EMA,2015)指出基于BCS的生物豁免标准必须应用于每种规格与对照药品的相应规格(EMA,2010;Cardot等,2016;EMA,2015)。美国FDA关于基于BCS生物豁免的指南草案(美国FDA,2015b)没有讨论在一系列规格的情况下需要哪种类型的数据来支持基于BCS的生物豁免。
本文的目的是研究EMA对其他规格生物豁免的各种要求,重点是仿制药产品上市的申请。
该综述还将比较EMA生物豁免要求和FDA的方法。这些比较将通过笔者基于日常实践设计的案例研究来说明。
2. EMA要求以及与FDA要求的比较
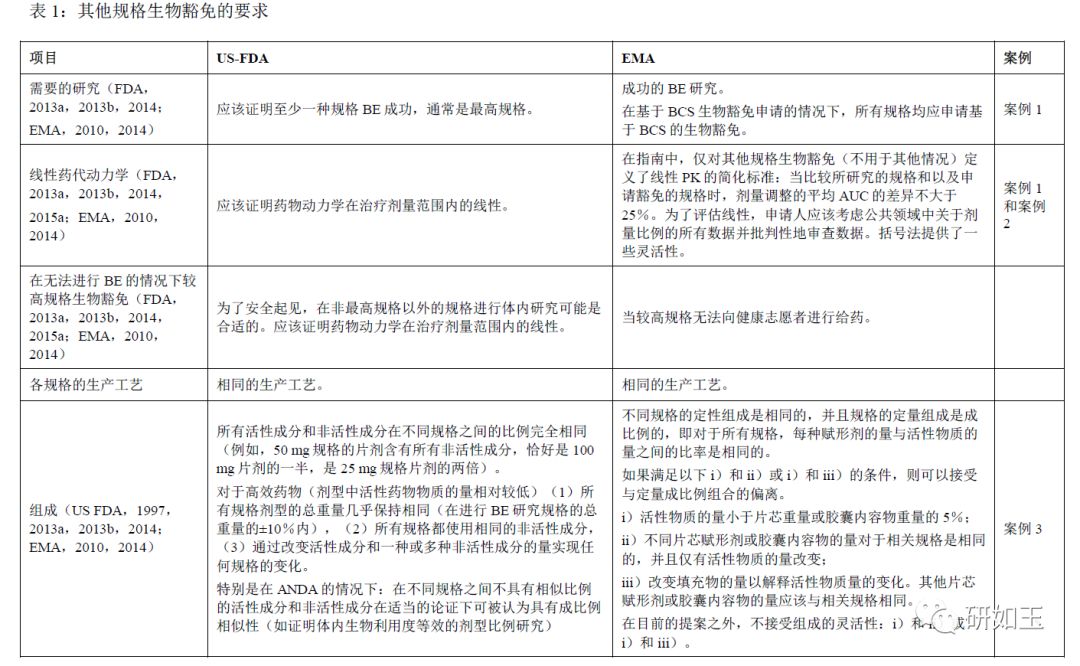
EMA指南(EMA,2010)于2010年更新,2013年、2014年、2015年(FDA,2013a,2013b,2014,2015a)发布了新的FDA指南草案。可以对其他规格生物豁免的要求进行对比。表1列出了关于获得其他规格生物豁免的主要EMA和FDA监管要求,涉及配方组成,需要证明体外可比性的证据,关于各种类型制剂的具体要求和其他信息。案例研究旨在展示这些概念如何应用于设计具有多种规格药品线的实际做法,并说明两个司法管辖区之间的相似性、困难和共同挑战如何影响药物制剂的开发。
3. 案例研究和讨论
3.1 案例1:药代动力学的线性和确定体内研究中使用规格的标准
3.1.1 EMA-FDA观点
应用其他规格生物豁免的先决条件之一是药代动力学呈线性。可以使用创新药公司EMA生成的数据评估药代动力学线性,以及公共领域获得的所有数据。这些数据必须经过严格审查。为了选择在BE研究中使用的规格,使用剂量调整的曲线下面积(AUC)评估线性,其应该满足±25%的标准(EMA,2010)。相比之下,美国FDA一般指南(FDA,2013a,2013b,2014,2015a)中没有提供类似的定义。
3.1.2 备注
剂量线性对选择进行体内临床BE研究的规格也发挥了作用。对于创新药具有线性药代动力学特性的速释(IR)制剂,通常选择较高的规格,除非存在安全性/耐受性问题,或药物高度可溶(EMA)。在这种情况下,体内研究可以选择较低规格。在剂量增加AUC呈比例增加的情况下,生物等效性研究一般应在最高规格上进行。在由于较低溶解度导致AUC不呈比例增加的情况下,应当在最高和最低规格上建立生物等效性。在不是由于较低溶解度(例如,转运蛋白的饱和)导致的AUC不呈比例增加的情况下,应当在最低规格或在曲线线性部分中的规格上建立生物等效性。
对于创新药具有线性药代动力学特性的调释(MR)制剂,研究数量和规格取决于药物产品是单一单位制剂还是多单位制剂,如表1和案例1所述。如果MR制剂呈现非线性药代动力学,那么用与IR产品相同的标准定义最灵敏的规格。由于MR制剂缓慢释放活性成分,由于转运蛋白或酶的较低溶解度或饱和度引起的非线性不会经常出现。
3.1.3 实例
实例1. 药物Y为具有4个规格的IR片剂产品线:5、10、20、40 mg。在10至100 mg间药代动力学描述为呈线性。因此,在40 mg规格在体内建立相对于对照药的BE。如果满足条件,可以应用其他规格的生物豁免,仅限于20 mg和10 mg,因为这两个规格在所述线性范围内。在EMA,无法授予5 mg规格的生物豁免,因为其超出了公开的范围。在这种情况下,一些国家可能需要对其他规格(5 mg)进行额外的BE研究。然而,如果较高规格呈现药代动力学线性不受任何饱和转运蛋白和代谢酶或溶解度限制,则可以假定最低规格也不会存在这些问题。因此,其他国家可能会接受5 mg规格体内测试的豁免。如有分歧,此问题可能需要在EMA-CHMP(人用药品委员会)进行仲裁。值得注意的是,FDA只规定必须在治疗剂量范围内证明剂量线性。如果没有数据,5 mg规格的可接受性问题是FDA的审评问题,除非产品特定指南中有公开明确指示。
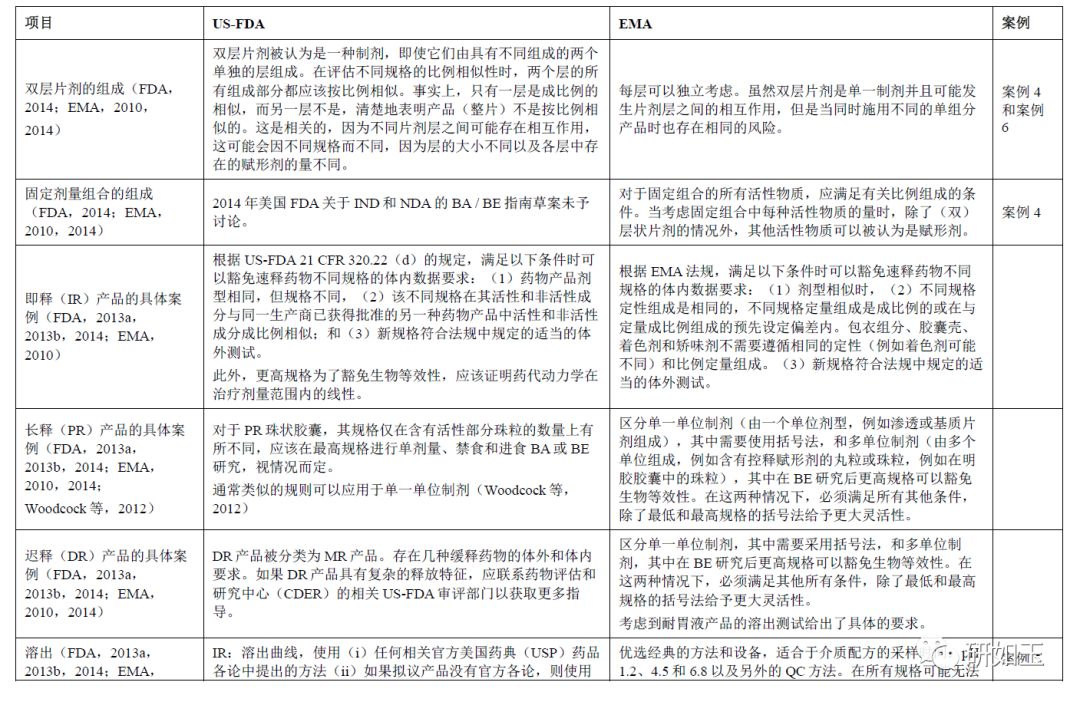
3.2. 案例2:支持其他规格生物豁免需要的体内数据
3.2.1 EMA-FDA观点
支持其他规格生物豁免所需的体内数据取决于配方是IR还是MR。如上所述,对于IR制剂,通常应在新产品线的最高规格上证明BE。相反,对于MR制剂,EMA区分长释(PR,在FDA指南中定义为缓释制剂:ER)和迟释制剂(DR),以及单一单位(例如片剂)和多单位制剂(例如胶囊壳内的丸粒/珠粒或压制成片剂)(EMA,2014)。
3.2.2 备注
在欧盟,单一单位制剂PR产品的情况下,通常应该以最高规格在三项体内研究中(如果没有安全性/耐受性问题)证明BE。根据创新产品的给药方案,这些研究在重复给药后积累的条件下,进行单剂量禁食、单剂量进食和多剂量禁食或多剂量进食(Paixão等,2012)。在单一单位制剂DR产品的情况下,多剂量研究不是必要的。另外,除了应用括号法外,还要求对每种规格进行一次单剂量体内BE研究。这个括号的界限应该是极端的规格(例如,最高和最低规格)或所有因素同时偏离豁免原则的极值(例如,两种规格在组成、溶出度或形状方面差异最大,所以括号限值涵盖剩余规格在组成或溶出度方面的任何差异)。最近欧盟的这一传统要求已被证明是必要的,因为在某些情况下体外溶出曲线不能确保BE(Woodcock等,2012;Lin等,2016)。在这种情况下,经典的生物豁免标准可以根据剂型的不同而增加一些特异性。对于PR制剂,测试产品所有规格的控释赋形剂和机制都应是相同的。DR制剂的控释包衣层也是如此。在多单位制剂的情况下,所有规格都必须使用相同的粒料。对于PR和DR,更高规格需要两个或三个BE研究;对于PR和DR以及另外对于具有稳态药物显著累积(AUC0-inf/AUC0-τ> 1.11)的PR,经典进食单剂量、禁食单剂量。如果像IR产品,生产工艺、组分和溶出要求符合要求,则较低规格可应用生物豁免。
在美国(美国FDA,2013a,2013b,2014,2015a),不论制剂释放机制的类型如何,必须进行至少两项研究:在进食和禁食状态下进行单剂量给药。基于FDA ER指南(美国FDA,1997),较低规格的豁免应符合几个标准,其中一个标准要求组分比例相似。如果较低规格的成分在比例上不相似,则根据所有规格进行的体内剂量比例研究或基于通常考虑最高规格和最低规格的体内括号法授予豁免。这突出了FDA与EMA的不同方法,就FDA而言,所要求的体内研究数量不是基于制剂类型,而是基于制剂组成(参见案例3)。
3.2.3 实例
实例2a. 药物Y为具有3个规格的基质片PR产品线:10、20和40 mg,其可以在无食物(或仅在禁食状态下)下服用。所有片剂的总重量为800 mg(赋形剂定性相同并且定量恒定在5%规则内),或者它们在数量上成比例并具有相同的形状。根据EMA指南,如果溶出曲线相似(假设药代动力学是线性的并且产品使用相同的工艺生产),则其他规格可以应用生物豁免。在这种情况下,应该在最高规格上进行禁食状态下的多剂量体内研究(假设药物积累)和进食状态下单剂量体内研究。另外,应该在最低和最高规格上进行禁食状态下的单剂量体内BE研究。
如果产品只能与食物共同给药(出于安全性或有效性的原因),则应在进食状态下进行单剂量最高和最低规格BE研究,在禁食状态下只进行最低规格的BE研究。并且应该在进食状态下进行多剂量研究,但高脂膳食不是必需的。
根据FDA关于仿制药产品BE研究的指导原则,如果组分被认为是按比例相似的,对照产品的药代动力学是线性的,不同规格使用相同生产工艺生产,且溶出曲线相似,则在最高规格进行两个单剂量BE研究是足够的。一项BE研究应在禁食状态下进行,另一项在进食状态下进行,除非出于安全原因仅在进食状态下给药。在后一种情况下,禁食状态下的BE研究可能对人类受试者不安全,并且在这种情况下,进食状态下的单剂量研究是足够的。
如果不满足5%的规则(例如,总片重恒定且),或者溶出曲线不一致但遵循规格之间的趋势,在欧盟则可以使用括号法。在这种情况下,三项体内研究必须在最低和最高规格上进行(6项研究),但如果不能满足组分和溶出条件,那么应该进行全部研究(9项研究)。
如果出现这种情况,鼓励申请人在产品开发期间联系相关部门寻求建议。
实例2b. 药物Y为具有3个规格的DR产品线:10、20和40 mg,以胶囊内的包衣小丸呈现。通过两项体内研究建立相对于对照产品的BE:最高规格进食和禁食单剂量(假设没有安全性/耐受性问题)。如果在每种规格中使用相同的药丸并且每个胶囊的药丸数量与规格成比例,则其他规格可以应用生物豁免。对于EMA和FDA都是如此。
3.3 案例3:制剂的组成
3.3.1 EMA-FDA观点
对于EMA和FDA,组成是在其他规格生物豁免中需评估的关键因素之一。
3.3.2 备注
如果医药产品采用相同的生产工艺生产,并且不同规格的定性组成相同,EMA描述了两种可能性:类似组成和5%规则(见表1)。
FDA通过以下两种方式之一定义比例相似性(美国FDA,2013a,2013b,2014):(1)不同规格之间所有活性和非活性成分的比例完全相同;或者(2)对于高效药物(剂型中活性药物物质的量相对较低),剂型的总重量对于所有规格应保持几乎相同(在进行生物研究规格的总重量的±10%内),所有规格都应使用相同的非活性成分,并且(美国FDA,1997)通过改变活性成分和一种或多种非活性成分的量实现任何规格的变化。特别对于仿制药产品的简化新药申请(ANDA)(FDA,2013a,2013b),如果不同规格之间活性成分和非活性成分在组成上不成比例,如果有充分的理由则可以认为成比例相似(如体内剂型比例研究证明BA等效)。
3.3.3 实例
实例3a. 药物X为具有3个规格的IR产品线:2.5、5和10 mg。通过在10 mg规格片剂的体内研究建立相对于对照药的BE。一家生产商开发出总重量为180 mg的片剂,API的减少被填充剂乳糖的增加所补偿。另一家生产商开发了具有相同API的2.5、5和10 mg规格的制剂,其中片剂总质量为205 mg并使用相同的配方策略,即API的降低通过增加填充剂(乳糖)来补偿。
两种情况下都可以使用规格生物豁免吗?
在欧盟,基于5%的规则,第一个重量为180 mg的配方不能获得其他规格生物豁免,因为对于5 mg和2.5 mg规格,活性物质的量小于片芯重量的5%,但不是生物研究规格10 mg(5.5%)。对第二个配方,基于5%的规则,可以豁免5 mg和2.5 mg规格的体内研究,因为对于10 mg(4.88%),5 mg(2.44%)和2.5 mg(1.22%)规格,活性物质的量小于片芯重量的%。FDA在确定哪种方法是合适的生物豁免方法时,两种情况下都会考虑API的性质(是否是高效药物物质)。考虑到药物以低剂量(10 mg或更少)给药,剂型的总重量对于所有规格保持相同(在BE研究规格的总重量的±10%内),所有规格都使用相同的非活性成分,任何规格的变化都是通过改变活性成分和一种非活性成分的量来获得的,根据其他充分理由,例如体内剂型比例研究证明了BA等效,这两种情况可能都被认为可接受。
实例3b. 药物X为具有4个规格的IR产品线:5、10、20和40 mg。通过在10 mg和20 mg规格片剂的体内研究建立相对于对照药的BE。其组成如表2所示。
表2:片剂中主要成分组成 | ||||
API或辅料 | 5 mg | 10 mg | 20 mg | 40 mg |
API mg (%片芯) | 5.00 (1.22%) | 10.00 (4.88%) | 20.00 (4.88%) | 40 (9.76%) |
填充剂 mg (%片芯) | 315.6 (76.98%) | 148.3 (72.34%) | 296.6 (72.34%) | 291.1 (71%) |
崩解剂 mg (%片芯) | 4.0 (0.98%) | 4.0 (1.95%) | 8.0 (1.95%) | 8.0 (1.95%) |
其他 mg (%片芯) | 85.4 (20.83%) | 42.7 (20.83%) | 85.4 (20.83%) | 79.9 (17.29%) |
片芯重量 | 410.0 | 205.0 | 410.0 | 410.0 |
申请人指出,基于在20 mg和40 mg上2次成功的体内研究,恒定的生产工艺和显示线性药代动力学产品的支持性溶出测试,5 mg和10 mg符合生物豁免的要求。申请人指出10 mg片剂是20 mg片剂(成比例组成)的1/2,并且5 mg片剂满足%规则,其质量与20 mg片剂相当。这个观点可以接受吗?
可以豁免10 mg规格的体内测试,因为它是20 mg规格的1/2,其在体内显示与对照药物生物等效。对于5 mg规格,满足5%规则,对于20 mg规格(可接受的BE研究),5 mg和20 mg规格片剂具有相同重量。然而,基于20 mg规格片剂对5 mg的生物豁免是不可接受的,因为活性物质量的差异应该仅由填充剂补充,而不是用崩解剂补充,其本应该是保持不变。因此,基于20 mg规格,最低规格5 mg的生物豁免是不可接受的。最低规格5 mg的生物豁免基于40 mg规格也是不可接受的,因为在比较规格时没有满足5%的规则。在这种情况下,申请人可以提交一份最低规格的新BE研究报告或从申请中撤回5 mg规格。相比之下,FDA可以接受5 mg规格的生物豁免,因为相对于20和40 mg规格,总配方重量是恒定的,并且仅通过两种非活性成分(填充剂和崩解剂)改变。在这些情况下,这些并不清楚,FDA建议申请人联系药物评估和研究中心的适当评估部门寻求建议。
3.4 案例4:固定剂量组合(FDC)
3.4.1 EMA-FDA观点
对于EMA,对于FDC药品,固定剂量组合中的所有活性物质应满足成比例组成的条件。当考虑固定组合中每种活性物质的量时,可以将其他活性物质视为“赋形剂”。在双层药片的情况下,可以单独考虑每层(EMA,2010,2014)。
FDA认为双层片剂是一种制剂,即使它们由两种不同成分组成的层组成(FDA,2014)。
3.4.2 备注
对于FDA来说,在评估不同规格的比例相似性时,两个层的所有组分均应该按比例相似。事实上,只有一层成比例相似而另一层不相似时可能表示药品(整片)不能成比例相似。这种方法考虑到不同片剂层之间可能存在相互作用,这可能会因不同规格而不同,因为层的大小不同以及各层中存在的赋形剂的量不同。
因此,EMA和FDA对于FDC片剂产品的生物等效豁免没有遵循相同的程序。
3.4.3 实例
实例4a. 假设对照FDC制剂由两种药物X和Y组成的单一IR片剂。在表3中列出测试片剂的组成以匹配对照制剂。所有其他生物豁免条件均视为满足。
表3:实例a FDC的组成 | ||||
组分 | 片1 | 片2 | 片3 | 片4 |
X(mg) | 4 | 4 | 8 | 8 |
Y(mg) | 5 | 10 | 5 | 10 |
辅料1(mg) | 80 | 80 | 160 | 160 |
辅料2(mg) | 80 | 160 | 80 | 160 |
总重(mg) | 169 | 254 | 253 | 338 |
根据与最高规格8/10 mg的成比例性,对4/5 mg规格可以豁免,但中间规格不能满足关于8/10 mg规格的任何定量组成方法。因此,应该对8 / 10 mg、8 / 5 mg和4 / 10 mg规格进行体内BE研究。
EMA指南提出了两种括号方法。这些选择是:以极端规格(例如最高和最低规格)或在组成差异最大的两种规格(API与赋形剂比例极端的配方)进行体内研究。极端规格为片1和片4,但它们是相似的,导致在BE研究中没必要对这两种片剂的给药进行研究。在组成上差异最大的两个规格为片2和片3。以百分比计,片3药物X(3.2%)和赋形剂1(63%)占比最高,赋形剂2(31.6%)和药物Y(2%)占比最低;片2药物Y(3.9%)和赋形剂2(63%)占比最高,药物X(1.6%)和赋形剂1(31.5%)占比最低。其余的片1和4在这些值之间。相反,两种药物最大数量一起代表片4。
根据申请人的策略,片2和片3可以在BE研究中给药,因为该括号法假定4/10和8/5的规格代表组成中的极端情况,没有进行最大量给药,即,8/10 mg,这不是极端情况之一。第二种选择是采用两个极端规格(片1和片4),但在这种情况下,两种配方是相似的,并且从配方差异的角度来看利益较低。
因此,确定了三种括号的极端情况。在最大限度的情况下,申请人应该对这三个规格进行BE研究(片2、3和4)。在中间情况下,申请人可以将BE研究限制在片2和片3中,认为这些片剂代表两种极端组成并且涵盖每种药物的单独规格。但是,一些欧洲国家可能会要求更多数据。这个例子说明了欧洲国家在指南解释上的差异。对于明确的指导可以通过EMA获得科学建议,尽管可能会收费。
第三种选择考虑到药物的量相对较小,这些配方应根据5%规则配制。应该配制8/5 mg规格以保持所有赋形剂不变(并且稀释剂可补偿药物Y中5 mg的差异或不像表4a和4b中所述)。类似地,对于4/10 mg规格,稀释剂可以补偿药物X中4 mg的差异。
表4a:实例a FDC的组成,满足规则(i)和(ii) | ||||
组分 | 片1 | 片2 | 片3 | 片4 |
X(mg) | 4 | 4 | 8 | 8 |
Y(mg) | 5 | 10 | 5 | 10 |
辅料1(mg) | 130 | 130 | 130 | 130 |
辅料2(mg) | 150 | 150 | 150 | 150 |
总重(mg) | 289 | 294 | 293 | 298 |
表4b:实例a FDC的组成,满足规则(i)和(iii) | ||||
组分 | 片1 | 片2 | 片3 | 片4 |
X(mg) | 4 | 4 | 8 | 8 |
Y(mg) | 5 | 10 | 5 | 10 |
辅料1(mg) | 121 | 116 | 117 | 112 |
辅料2(mg) | 150 | 150 | 150 | 150 |
总重(mg) | 280 | 280 | 280 | 280 |
表4a和4b中提出的两个配方与规格生物豁免的规则一致,但最终片剂的质量和大小与创新药的片剂配方大不相同。这可能是FDA(US FDA,2015d)关注的问题,FDA现在建议:“如果RLD(参比制剂)的最大尺寸小于17 mm,那么仿制产品任何单一维度应不超过RLD的20%(仿制单个维度结果不应超过17 mm),并且不超过RLD体积的40%。如果RLD的最大尺寸等于或大于17 mm,则仿制产品任何单一维度不得大于RLD体积(美国FDA,2015d)。在目前的情况下,对于较低规格,与对照(表3)相比,质量增加65%,这对于FDA批准生物豁免可能是个问题。
实例4b. 两种药物X和Y,现在配制为IR双层片剂。片剂的组成列于表5中。
表5:实例4b FDC的组成 | ||||
组分 | 片1 | 片2 | 片3 | 片4 |
层1 API X(mg) | 4 | 4 | 8 | 8 |
层1 辅料(mg) | 80 | 80 | 160 | 160 |
层2 API Y(mg) | 5 | 10 | 5 | 10 |
层2 辅料(mg) | 80 | 160 | 80 | 160 |
总重(mg) | 169 | 254 | 253 | 338 |
每层中药物与赋形剂的比例是恒定的,并且可以认为该规则满足EMA,因为可以单独考虑每一层。在目前的情况下,EMA认为双层片剂的摄入量与两个独立单组分片剂对照产品的同时摄入相当。如果参比片剂的组成成比例,则其在胃肠液中崩解和混合后对应于双层片剂的每层。
然而,对于FDA而言,双层片剂被认为是一种制剂,即使它们由具有不同组成的两个独立层组成。在评估不同规格的比例相似性时,两层的所有组分均应该按比例相似。美国拒绝生物豁免申请。
实例4c.两种药物A和B,在单一单位IR片剂中呈现。片剂的组成列于表6中。
表6:实例4c FDC的组成 | ||||
组分 | 片1 | 片2 | 片3 | 片4 |
API A(mg) | 10 | 20 | 20 | 40 |
API B(mg) | 5 | 5 | 10 | 10 |
辅料(mg) | 185 | 175 | 370 | 350 |
总重(mg) | 200 | 200 | 400 | 400 |
所有规格的API与赋形剂的比例都不是恒定的。存在两组:40/10和20/5与20/10和10/5。在这种情况下,根据EMA和FDA的规定,应该进行两项体内BE研究,并且如果满足其他条件,另外两个规格可以获得生物豁免。
3.5 案例5:溶出度测试
3.5.1 EMA-FDA观点
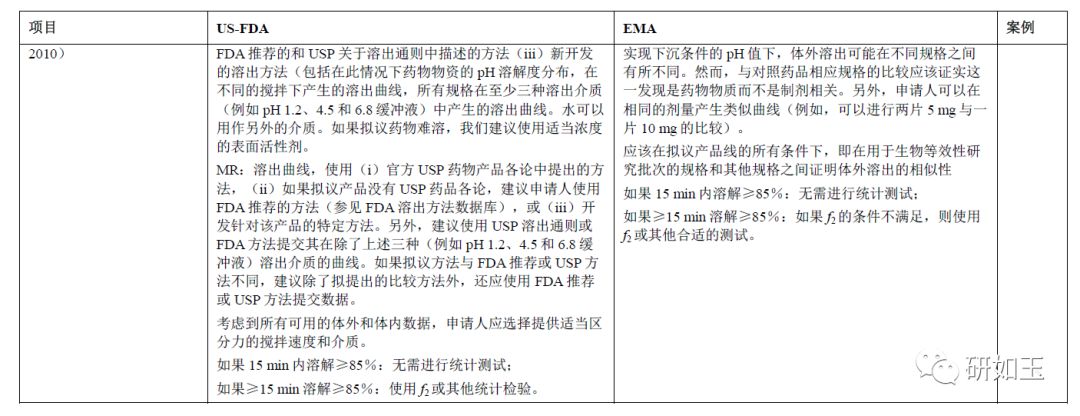
FDA和EMA都要求进行溶出度测试,以支持其他规格的生物豁免。对于EMA,它们基于使用1.2、4.5、6.8的3种常规pH介质和质量控制(QC)方法的标准化测试。对于FDA,溶出度测试应使用USP各论进行(如果有的话)。如果没有各论,则使用FDA推荐的监管方法。如果既没有USP也没有合适的FDA方法可用,则要求申请人开发针对该产品的特定方法。在后一种情况下,FDA还要求申请人使用3种常规pH值介质1.2、4.5和6.8提供体外溶出度测试(FDA,2013a,2013b,2014;FDA,2017b;EMA,2010)。
3.5.2 备注
EMA支持其他规格生物豁免所需的溶出度测试是基于3种常规pH为1.2、4.5和6.8的介质。如果不同,还应该使用基于QC方法进行第四次测试。即使药物是难溶性药物,pH 1.2、4.5和6.8的介质也不得加入任何表面活性剂。只有QC介质可能含有表面活性剂。明确的是,在所有规格均无法达到下沉条件的pH值下,可以接受不同规格的体外溶出不同。然而,与对照药品相应规格的比较应该证实这一发现与药物相关,而不是与配方相关。或者,即使测试和对照产品在每个规格水平上都没有表现出相似的溶出曲线,申请人也可以在每个容器相同剂量下显示类似的曲线(例如,可以在一个容器中比较两个5 mg片剂的溶出度与在一个容器中一个10 mg片剂)。应该在所应用的产品线内的所有条件下,即在用于体内BE测试批次的规格和其他规格之间,证明体外溶出的相似性。因此,对于EMA,与对照产品的体外比较不是强制性的。
在美国(FDA,2014年指南草案),每种规格的溶出曲线都应使用推荐的QC监管溶出方法生成。如果尚未最终确定监管溶出方法,则应在至少三种介质(例如pH 1.2,4.5和6.8)中生成溶出曲线。
对于曲线比较,当剂型中> 85%的药物标示量在溶解时,不需要定量统计方法来比较溶出曲线。否则,可能需要f2测试来比较两种溶出曲线。当仅由于可变性原因使用f2进行平均曲线比较不适用时,应使用替代曲线比较测试。尽管美国FDA现行指南中没有充分说明关于所有可能替代测试的监管接受程度以及计算可接受限度方法的详细信息,替代测试可以考虑自举f2相似因子和多变量统计差异(MSD)90%置信区间(EMA,2010;FDA,2013a,2013b;Cardot等,2017;Mangas-Sanjuan等,2016;Paixão等,2017)。假设曲线相差10%时,可以根据获得的MSD定义可接受范围。
在美国,根据仿制药法律,每种仿制药都需要对照(“橙皮书”中列出的“参比制剂”),因此有必要对每种产品的所有规格进行体外溶出度测试(测试和对照)。然而,FDA认为测试–对照体外溶出曲线可能不匹配,因为美国法律允许仿制药与参比药物具有不同的配方。因此,可能不一定有f2测试–对照溶出曲线比较,但是仿制药申请人仍然需要提交一套完整的测试和参比溶出曲线。FDA将评估产品线中仿制药产品所有规格的溶出曲线相似性。
3.5.3 实例
实例5a. 规格为25、50和100 mg的软IR胶囊由软明胶壳内的液体混合物组成。胶囊填充剂量是成比例的。体内BE研究在100 mg规格下是可接受的,并且对照产品表现出药代动力学剂量比例性。由于API显示较低溶解度,溶出QC方法使用USP设备2(桨),转速为75 rpm,在1000 mL含有0.5%月桂基硫酸钠的0.1N HCl中,37 oC。在没有表面活性剂的情况下,在pH 1.2、4.5和6.8下的溶出由于再沉淀而可以忽略,因此在各规格上相似。在具有表面活性剂的QC介质中溶出/崩解期间,与对照产品类似,在10 min内三种规格显示> 85%溶解。三种规格的崩解(或破裂)时间均。
从化学制药的角度来看,如果证明这些溶出条件具有足够的区分性(有问题的问题可能是1000 mL的大容量介质,以及向介质中添加表面活性剂),则生物豁免是可以接受的。在溶出方法开发和验证期间,使用具有关键工艺参数和/或可能对生物利用度有影响的关键物料属性差异的批次建立区分力。例如,可以改变所用赋形剂的比例或改变生产工艺以举例说明溶出测试的区分力。对美国而言,最好是申请人提供证据表明诸如此类的溶出方法具有充分的区分力。值得注意的是,FDA希望申请人应该常规使用USP或FDA溶出方法(FDA,2017b)(如果已发布)。然而,公布的溶出方法可能并不总是适合仿制药。这是因为公布的溶出方法通常对应于针对特定医药产品开发的QC方法,并且许多因素可能在新配方中相互作用,例如API来源、配方和生产工艺(Scheubel等,2012)。在这种情况下,申请人应提交证据表明,与USP或FDA溶出方法相比,不同的溶出方法提供了最佳的区分力。
实例5b. 药物X为IR配方产品线,具有3个规格:10、20和40 mg。通过对40 mg的体内研究建立相对于对照品的BE。配方是相似的并且产品表现出药代动力学线性。该药物表现出较低溶解度,其导致pH和规格依赖性溶出。在pH> 6的介质中,溶出非常好。在pH = 1.2时,最低规格的溶出度达到最高8%的平台,而40 mg规格达到平台约2%。在pH 4.5的介质中,较低规格达到100%溶解,最高规格只有25%溶解。在pH 6.8(QC介质)中,所有规格在内溶解> 85%。
对于EMA,在pH 1.2时,由于溶解量较低(规格之间的差异不能大于10%),因此不需要统计溶出曲线比较测试(因为溶解最大量%,2%和8%)。在pH 4.5时,由于规格依赖性溶出,各规格显示不同的溶出曲线。考虑到每个容器的类似剂量,即40 mg,可以用第二组溶出测试来解决该问题。在实践中,应将1×40 mg片剂与2×20 mg片剂和4×10 mg片剂进行比较。然后溶出必须相似,这表明它是与药物有关的溶解度问题,而不是配方差异。此外,测试和对照在pH的行为必须与溶解性差以及随后溶出差相似。这种在容器中使用相同剂量来比较难溶性药物各种规格的方法在美国FDA尚未标准化(Suarez-Sharp等,2016)。
实例5c. 药物X在欧洲被配制为IR产品线,具有3种规格:10、20和40 mg。通过用40 mg规格(单剂量禁食)进行的体内研究建立相对于对照产品的BE,因为对照产品可以与或不与食物共同服用。配方是相似的并且产品表现出药代动力学线性。当介质中不加入表面活性剂时,药物几乎不溶。QC介质由pH 6.8 + 2%吐温80组成。
基于药物的较低溶解度,申请人证明没有必要研究没有表面活性剂的情况下在缓冲介质中的溶出,因此申请人不提供pH 1.2、4.5和6.8中的任何溶出数据。这个建议可以接受吗?EMA的答案是否定的,应提供在所有介质中的的溶出。
随后,申请人在每个试验中使用6个单位(每个容器1个单位)在pH 1.2、4.5和6.8提供有限的溶出度数据。这可以接受吗?答案是否定的,应提供12个单位的溶出曲线。然而,一些欧盟国家可以同意,不需要对12个单位进行论证,因为需要12个单位才能进行正式比较,但有6个单位足以证明没有表面活性剂的溶出是可以忽略的。在美国(FDA,2013a,2013b准则草案),应使用推荐的溶出方法生成每种规格的溶出曲线。如果尚未最终确定监管溶出方法,则应在至少三种介质(例如pH 1.2、4.5和6.8)中生成溶出曲线,优选12个单位。
实例5d. 药物X在欧洲被配制为IR产品线,具有3种规格:10、20和40 mg。通过用40 mg规格进行体内研究建立相对于对照产品的BE。申请人决定比较每种规格的测试和对照产品的体外溶出度以支持其他规格生物豁免。足够吗?这是不够的,对于EMA,申请人应在测试产品的产品线内提供其他规格与BE研究中所研究规格的比较。在每种规格水平下与对照产品的比较仅在测试产品不同规格没有由于缺少漏槽条件而显示类似曲线的情况下才是具有支持性的。FDA认为体内BE是可以接受的,通常期望看到对于测试产品和对照产品每种规格的完整溶出曲线集合,虽然对于曲线相似性的统计比较只应在测试产品线内进行,并与体内BE研究中测试规格的溶出性能进行比较。
3.6 案例6:双相双层片剂
3.6.1 EMA-FDA观点
在EMA指南中,双层片剂被描述为配制FDC药物产品的一种选择。在FDC中,每一层都可以独立考虑。根据美国FDA指导原则,双层片剂被认为是一种配方,即使它们由两种不同成分组成(EMA,2010,2014;FDA,2014)。
3.6.2 备注
在评估由于长释层和快释层而显示双相体外释放双层片剂的情况下,没有明确建立规则。案例6为这种情况提供了一种可能的情景。
3.6.3 实例
实例6. 药物X为MR制剂产品线中的两种规格:40和50 mg,其包含10或20 mg的快释层和30 mg的迟释层(10 / 30 mg或20 / 30 mg)。通过使用50 mg(20 / 30 mg)规格的三项BE研究建立相对于对照产品的BE。IR层在数量上是成比例的,并且PR层在所有规格上是相同的。申请人支持具有相对溶出度的10/30 mg规格的生物豁免,因为PR层是相同的并且只有IR层改变。这种方法可以接受吗?如果申请人证明这些层的溶出曲线相似,则可以认为PR层与IR层没有相互作用。因此在欧盟这种方法是可以接受的。
相反,FDA认为双层片剂是一种配方,即使它们由具有不同组成的两个独立层组成。在评估不同规格的比例相似性时,两个层的所有组成部分均应该按比例相似。事实上,只有一层比例相似而另一层不是比例相似,并不意味着FDA会断定整个药片的比例相似。FDA认为这是相关的,因为担心不同片剂层之间可能存在潜在的相互作用,这可能会因不同规格而不同,因为层的大小不同以及各层中存在的赋形剂的量不同。
3.7 案例7 新规格
3.7.1EMA-FDA观点
对于EMA,如果满足所有先决条件,则可以在产品特性摘要(SmPC)所接受的治疗范围内接受其他规格的生物豁免。这意味着如果最高规格和最低规格在体内与对照产品相当,那么中间规格也可以被接受。这种方法的积极结果是患者不需要破坏最高规格的片剂或服用两片最低规格的片剂来达到合适的剂量。
3.7.2 备注
根据EMA或FDA的指导方针,使用相同的方法不能清楚地解决某些问题;例如,为填补可用给药方案的缺口以改善患者依从性而开发/引入一种新的市场上没有的规格。实例7a和7b中描述了这种情况。
3.7.3实例
实例7a. 药物X为IR配方产品线,具有3种规格:10、20和40 mg。通过对40 mg的体内研究建立相对于对照产品的BE。10 mg是一种具有划线的易碎片剂,因此可以产生1/2片剂,两片相当于5 mg剂量。5 mg剂量在SmPC中被描述为可能的治疗方法,例如用于(i)在剂量递增期间开始治疗直至达到治疗剂量,(ii)在一些具体的适应症中,具有较低剂量或较低体重患者的剂量调整或肾/肝损伤。申请人决定配制一个5 mg片剂,以避免掰片,并改善患者的依从性,特别是考虑到老年患者的需要(更简单易用)和降低成本的可能性(在医院不保留半片药片)。5 mg片剂与10 mg片剂的成分成比例,并未由原研厂家销售。在EMA,为了支持5 mg规格的生物豁免,申请人仅需要开发5 mg规格成比例组分或在5%规则的要求范围内,并显示5 mg规格和体内研究的40 mg规格之间相同的溶出曲线。没有必要进行5 mg规格与10 mg片剂的一半或2×5mg片剂与1×10 mg片剂的比较。不能认为这种新规格是仿制药,因为对照5 mg规格并未上市,但其可以以混合申请进行提交。相比之下,美国FDA允许新规格在适用性请愿书中作为仿制药进行开发。适用性请愿书要求允许提交ANDA,以便针对某些特征(如本案例中的规格)与列出的药物不同的药品。根据21 CFR 314.93(e),FDA将在提交适用性请愿书后不迟于90天批准或否决适用性请愿书(FDA,2013a,2013b)。如果适用性请愿书获得批准,那么仿制必须在美国提交适当的上市许可研究;最有可能的是一项体内研究,其中8×5 mg的新规格必须与40 mg体内生物等效。如果FDA接受BE研究,那么这种新5 mg规格将成为任何未来仿制5 mg规格的参比(但仅限于该规格)。
实例7b. 药物X为IR配方产品线,具有3种规格:10、20和40 mg。通过对40 mg的体内研究建立相对于对照产品的BE。SmPC表明最大剂量是2片40 mg。由于SmPC包含了将两片最大规格的片剂组合在一起的可能性,有可能开发这种规格。如果动力学是线性的并且根据BCS标准药物高度可溶,则可以基于溶出数据在欧盟获得批准。如果其呈现较低溶解度,则有必要将新规格与两片对照产品当前最高规格(40 mg)进行体内比较。
通过适用性请愿书途径,可以在美国提交用于上市这一新规格的申请(FDA,2013a,2013b)。适用前一个例子中新5 mg规格相同的要求。在这种情况下,如果适用性请愿书获得批准,那么要求仿制药发起人进行1×80 mg(新)与2×40 mg(对照)的体内研究。80 mg将成为任何未来发起人拟上市80 mg规格的新参比。然而,随后的仿制药仍然有必要在40 mg规格上进行体内研究,因为这用于III期临床试验,并且是批准的基础。最有可能的是,如果80 mg被批准,仍然需要进行两次40 mg规格体内研究。
相反,如果创新药品的剂量说明中没有描述新的最高或最低规格,那么开发这些新规格将需要临床研究来证明新拟议剂量的有效性和安全性。
4. 结论
在仿制药开发中,其他规格生物豁免是一种用于IR和MR剂型的非生物研究规格非常成熟的方法。FDA和EMA指南都提供了一些关于如何建立其他规格生物豁免的具体建议,这些生物豁免可能与指南的出版日期或当地的特殊性和规定相关。在研究EMA与FDA方法比较评估的监管指南时,我们总体指出,EMA指南倾向于为FDA指南的某些情况提供更具体的建议。在美国,鼓励申请人联系药物评估和研究中心的适当临床审查部门,以获得产品开发期间的建议(FDA,2015c)。FDA不会通过电话沟通、书面交流或面对面的会议向发起人提供监管建议的收费。相反,EMA提供的科学建议需要收费。FDA(2014年)指南草案与EMA关于如何申请和获得生物等效性的指导方针之间的融合很显著,但这一领域仍然存在许多差异。可以得出结论,FDA在配方组成差异方面比EMA更灵活,除了双层片剂的情况;而EMA需要更多溶出度比较。对于PR产品,由于要求最低规格单剂量BE研究,不同于通常要求进行最高规格体内研究的FDA(除非出于安全考虑),因此EMA不允许在单一单位制剂的情况下使用一套完整的生物豁免降低产品线。最后,值得注意的是,在欧盟预计仿制药和创新药将遵循类似的原则来推导生物豁免。
参考文献
略
© 版权声明
文章版权归作者所有,未经允许请勿转载。
相关文章




暂无评论...