文章来源:研如玉
编者语:临床试验失败很常见,成功的都是少数,本文就这个问题进行了细致的分析并给出了提高临床试验成功率的一些有意义的建议。本文来源见以下截图:

为什么90%临床药物研发失败?要如何提高临床试验的成功率?
Duxin Suna,*,
Wei Gaoa, Hongxiang Hua, Simon Zhouba密歇根大学药学院药学系, 安娜堡, MI, 48109, USAb转化开发和临床药理学,百时美施贵宝公司,Summit,NJ,07920,美国2021年 11 月 20 日收到; 2022 年 2 月 3 日以修订形式收到; 2022 年 2 月 6 日接受摘要
尽管实施了许多成功的策略,但90%的临床药物开发都失败了,这引发了一个问题:是否忽略了靶点验证和药物优化的某些方面?当前的药物优化过度强调使用结构的效力/特异性。但选择相关性(SAR)忽略了使用结构的疾病/正常组织中的组织暴露/选择性。组织暴露/选择相关性(STR),可能误导候选药物的选择并影响临床剂量/疗效/毒性的平衡。我们提出结构。组织暴露/选择性活性关系(STAR)用于改进药物优化,它根据药物的效力/选择性、组织暴露/选择性以及平衡临床疗效/毒性所需的剂量对候选药物进行分类。I类药物具有较高的特异性/效力和较高的组织暴露/选择性,需要低剂量才能获得较高的临床疗效/安全性和较高的成功率。II类药物具有较高的特异性/效力低组织暴露/选择性,需要大剂量才能达到高毒性的临床疗效,需要谨慎评估。III类药物具有相对较低(足够)的特异性/效力,但组织暴露/选择性较高,这需要低剂量才能达到毒性可控的临床疗效,但往往被忽视。IV类药物特异性/效力低,组织暴露/选择性低,其疗效/安全性不足,应尽早终止。STAR可以改进药物优化和临床研究,以实现临床药物开发的成功。关键词:药物开发;药物优化;临床试验;结构组织暴露/选择相关性(STR);结构组织暴露/选择活性关系(STAR)
2022 中国药学会、中国医学科学院药物研究所。由Elsevier B.V. 制作和托管这是 CC
BY-NC-ND 许可下的开放获取文章。1.为什么90%的临床药物研发失败?
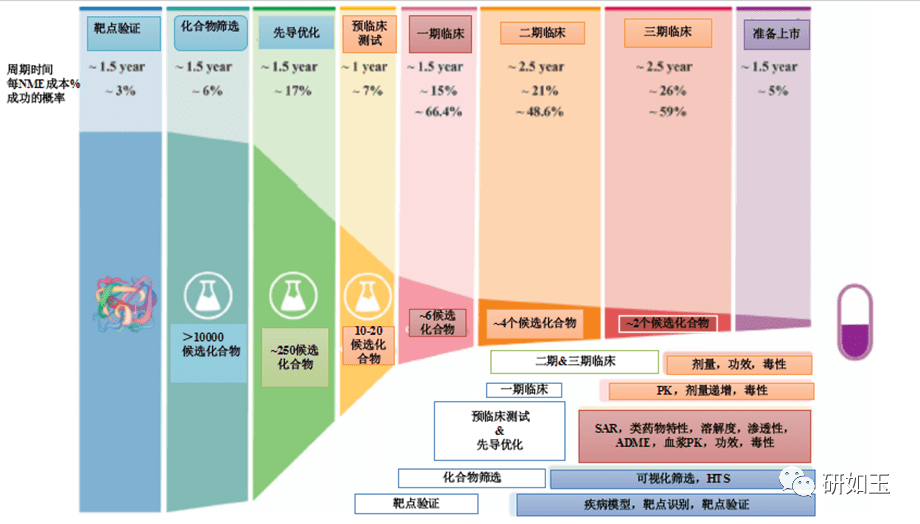
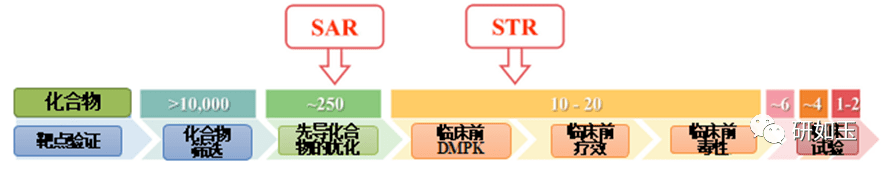
药物发现和开发是一个漫长、昂贵和高风险的过程,需要超过10 到15 年的时间,每种新药获批临床使用的平均成本超过1 到20 亿美元1。对于任何制药公司或学术机构来说,在临床前阶段对候选药物进行严格优化后,将候选药物推进到I期临床试验是一项重大的成就。然而,在进入临床研究后,十分之九的候选药物会在I、II、III 期临床试验和药物批准过程中失败2、3。还值得注意的是,90%的失败率是针对已经进入I期临床试验的候选药物,这不包括处于临床前阶段的候选药物。如果把临床前阶段的候选药物也算在内,药物发现/开发的失败率甚至高于90%。对2010 年至2017 年临床试验数据的分析表明,90%的药物开发临床失败可能有四个原因:缺乏临床疗效(40%-50%)、难以控制的毒性(30%)、不良的药物性能(10 %-15%),以及缺乏商业需求和糟糕的战略规划 (10%)2,4。一般来说,药物开发遵循一个经典过程(图1),包括严格的基因和基因组靶点验证、候选药物分子的高通量筛选(HTS)、严格的药物活性和类药物性质优化、临床前疗效和毒性测试,生物标志物指导的患者选择和最佳临床试验设计。在过去的几十年里,上述药物开发的每一步都经过了严格的优化和验证,同时在药物开发过程中,许多成功的策略也得到了正确的实施,以选择临床研究的最佳候选药物。尽管进行了这一验证,临床药物开发的总体成功率仍然很低,为10%-15%5-7。如此持续的高故障率引发了几个问题:尽管过去几十年实施了许多成功的策略,为什么90% 的临床药物开发却失败了?我们是否忽略了导致高失败率的药物开发过程的某些方面?如何提高临床药物开发的成功率?
图1药物发现和开发的过程,以及每一步的失败率。
2.在过去几十年中,有哪些成功的策略可以改善药物开发过程的各个方面?
2.1. 选择最佳候选先导药物,以达到足够的临床疗效
由于40%-50% 的药物开发临床失败是由于缺乏临床疗效,因此在临床前和临床研究中,人们一直致力于提高药物疗效。在目标验证过程中,通过对细胞系、组织、临床前模型和人类疾病模型5、7-12 中的遗传学、基因组学和蛋白质组学研究,严格确认了疾病目标。然而,由于体外、疾病动物模型和人类疾病之间的生物学差异可能会阻碍对分子靶点功能的真正验证,因此在成功开发药物之前,对人类疾病中任何新分子靶点的真正验证都是一个挑战8-12。这种差异使一流药物的开发变得困难。在药物筛选过程中,通常同时进行虚拟计算筛选和化学库的 HTS,以选择最佳支架并消除与靶标的非特异性结合6、7、12、13。人工智能 (AI) 和机器学习计算工具进一步改进了计算辅助药物设计过程14,15。HTS 使用基于蛋白质的生化测定、基于细胞的表型分析或基于生物体的分析,可提高hits6、7的效率和特异性。在药物优化过程中,先导化合物通过构效关系(SAR) 进行广泛优化,以实现对其分子靶标的高亲和力和特异性(Ki或IC50 在低nmol/L 或pmol/L 范围内)并限制脱靶效应16
-20。然而,药物分子的药理作用的验证是药物抑制其预期分子靶标的结果,这也可能具有挑战性,因为该分子的药理作用(功效和毒性)可能是由于抑制了一些未知的分子靶标12.有趣的是,一些药物已经成功开发,尽管它们的目标与之前报道的预期目标不同12。然而,在这种情况下,使用SAR 对预期目标进行药物优化可能会误导优化工作。在临床前测试期间,通常对化合物进行测试和优化,以在临床前动物疾病模型中显示出优异的功效。已经建立了许多动物模型来模拟人类疾病状况,尽管找到最佳动物模型来完全重现人类疾病的疾病表型或病理生理学仍然是一个挑战5。在临床研究过程中,从I期到III期的临床试验设计和剂量方案选择都经过严格优化。此外,基因组学和遗传生物标志物也被广泛用于选择患者进行临床试验,以提高药物开发的成功率21。值得注意的是,大部分药物发现工作(在临床研究之前)都正确地投入到这些研究中,以选择具有与人类疾病相关的有效分子靶标的最佳候选先导药物。然而,在临床I、II和III 期研究中,许多“完美”候选药物仍有很大比例失败。尽管上述药物发现努力是有合理的,但过分强调上述努力(即目前药物发现的实践)可能会有所帮助,但可能无法解决临床药物开发持续的高失败率问题。2.2 选择最佳的先导候选药物以最大限度地减少临床毒性
毒理学研究中使用了各种策略,以尽量减少由于无法控制的临床毒性22,23而导致的药物研发失败。药物的毒性可能由分子靶点的非靶向或靶向抑制引起。为了减少非靶向毒性,通常会针对其他靶点进行药物筛选24,25。例如,开发任何激酶抑制剂通常需要针对其他数百种激酶进行筛选。疾病相关激酶靶点与其他激酶靶点的选择性通常通过其IC50的比率来计算,其中优选至少10倍的选择性。另一方面,如果候选药物具有靶向毒性,这是由抑制疾病相关靶点引起的,那么在滴定剂量方案可能有帮助的情况下,解决方案是有限的。此外,候选药物通常会被评估是否会抑制主要重要器官中的几个已知毒性靶点。例如,体外和体内hERG检测通常作为先导化合物心脏毒性(致命扭转性心律失常)的预测指标26。最后,候选药物也可能在没有明确已知靶点的情况下引起化学诱导毒性。毒理基因组学通常用于潜在毒性的早期评估14,27。候选药物的结构可能需要进一步修改,以尽量减少药物–蛋白质加合物或药物-DNA加合物,从而降低潜在的器官毒性28。体外/体内动物研究总是针对潜在的遗传毒性和致癌性22。实际上,候选药物的急性和慢性毒性(模拟临床剂量方案)总是在三种动物身上进行检测23。由于动物模型和人类之间可能存在差异,在临床试验期间,通常使用各种方法优化剂量方案,以维持具有足够疗效和可控毒性的治疗窗口23。然而,无论候选药物对分子靶点有非靶向或靶向毒性,候选药物在重要器官或血细胞中的累积是导致毒性的主要因素之一。不幸的是,目前还没有完善的策略来优化候选药物,以减少主要重要器官中的组织积聚,从而将潜在毒性降至最低。2.3 选择具有最佳类药物特性的最佳先导药物候选
20世纪90年代,由不良的药物性质造成的药物研发失败占30%-40%;但它们只占当今药物研发失败的10%-15%29。这种改进得益于药物优化过程中对类药物性质的严格选择标准,包括溶解度、渗透性、蛋白质结合、代谢稳定性和体内药代动力学,如生物利用度(F)、药物暴露(AUC)、Cmax、t1/2、清除率CL和体积分布V30。这些类药物特性的某些临界值已被用作选择最佳先导化合物的标准。在化学结构设计中考虑了“5法则”:(1)少于5个氢键供体;(2)分子量小于500;(3) cLogP小于5;(4)少于10个氢键受体31。还需要小于140 A2的极性表面积32。为了更好的口服吸收,优选超过2×10-6~3×10-6cm/s体外渗透性33。根据生物药剂学分类系统34,35,进行预制剂和制剂研究,以增加药物溶解度并提高口服生物利用度。优选体外微粒体稳定性t1/2>45-60分钟。同时,严格执行临床前药代动力学,筛选出生物利用度F >30%、半衰期t1/2 > 4-6 h、清除率CL 肝血流量Q 的合适类药性质的先导化合物36,37。值得注意的是,在过去20年中,由于类药物性质差而导致的临床药物研发失败得到了显著改善29,这表明了类药物性质优化的正确策略。然而,临床药物开发的总体成功率并没有显著提高,仍处于10%-15%的低水平。然而,尽管对药物性质的所有体外和体内评估都得到了充分发展和充分证明,但目前的选择标准使用了血浆药代动力学,这可能会或可能不会指导临床试验中正确选择主要候选药物。通常血浆暴露较好的候选药物会被选择进入临床研究,而血浆暴露较低的候选者通常会在没有进一步开发的情况下被淘汰38,39。然而,候选药物的临床剂量/疗效/毒性取决于疾病靶器官与正常器官中的有效药物暴露/选择性,但当前候选药物选择中的药物优化过程尚未充分利用标准来评估疾病靶器官与正常器官中的药物暴露/选择性,这可能误导了候选药物的选择40。药物血浆暴露可用作疾病靶向组织中治疗暴露的指标,这一假设是基于“游离药物假设”来选择先导候选药物。该假说认为,只有血浆中游离的未结合药物(而非血浆蛋白结合药物)才能分布到疾病靶向组织中与其分子靶点相互作用;而血浆中的游离药物暴露与稳态时的疾病靶向组织相似41、42;因此,血浆中的药物暴露可用于预测候选药物的药效学作用。然而,这种“游离药物”假设可能仅适用于有限类别的候选药物,但不适用于许多其他化合物,因为许多因素会导致血浆和组织之间的游离药物分布不对称42-64。因此,在不了解疾病靶向组织/正常组织中的药物暴露情况下,血浆中的药物暴露可能会误导临床试验候选药物的选择51-53,65。2.4 优化药物开发战略规划
不良的战略规划(可能包括治疗重点的改变、公司合并或不良的临床研究行为)占药物开发失败的 10%2。制药公司的合并可能会增加被迫终止的药物重复数量2,4. 所有制药公司都制定了细致的开发计划,并制定了详细的路线图和里程碑,以推动新化合物从实验室通过每个开发阶段。由经验丰富的专家组成的多学科项目团队经常在各种商业模式和分析工具的帮助下共同制定战略规划66。人工智能 (AI) 带来了最先进的分析工具,使制药公司能够以更有效和更具成本效益的方式预测患者的需求和市场趋势14。3.药物发现/优化中哪些被忽视的方面导致临床药物开发的高失败率?
3.1真正的靶点验证,即确认分子靶点是人类疾病的原因和药物的预期靶点,对于临床药物开发的成功仍然具有挑战性
任何药物发现和开发计划都需要对两种类型的靶点验证进行严格研究8,9,16-20。一种类型的靶点验证是确认分子靶点确实是人类疾病的原因。尽管在体外细胞系、动物疾病模型和人类疾病模型中使用各种遗传或基因组方法对这种类型的靶点验证进行了广泛研究,但分子靶点的生物学差异仍然存在于体外和体内或动物和人类疾病之间10。事实上,在开发出成功的药物之前,不会对分子靶点进行充分验证,这给一流的药物发现和开发带来了挑战。另一种类型的靶点验证是确认分子靶点是否是药物分子的预期靶点,这通常通过SAR研究结合分子靶点的特异性/亲和力来确认16-20。药物分子的药理作用可以与使用siRNA敲除或CRISPR基因编辑对靶标进行基因改造的效果11,12进行比较。然而,药物靶点的验证仍然具有挑战性,因为药物的药理作用(功效和毒性)可能是由于对可能与其预期靶点不同的未知分子靶点12的抑制而产生的,从而影响如下所述的药物优化过程。有趣的是,一些药物已经成功开发出来,尽管它们的靶点与之前报道的预期靶点不同12。事实上,这两种靶点验证仍然存在许多挑战,如前所述,这可能会导致临床药物开发的高失败率8,9。然而,即使目标确实得到验证,许多候选药物在临床I、II 和 III 期试验中仍然存在很高的失败率,这凸显了如下所述候选药物优化过程的重要性。3.2当前的药物优化可能过分强调了一个方面,而忽略了可能误导候选药物选择并影响临床剂量/疗效/毒性平衡的其他方面
3.2.1当前药物优化流程需要对先导候选药物进行两方面的优化
在药物优化过程中,严格优化了化合物的两个主要方面:(1)通过SAR严格优化先导化合物抑制分子靶点的效力和特异性,需要低nmol/L甚至pmol/ L范围内的低Ki或IC50,达到更好的疗效并减少脱靶效应; (2) 使用某些临界值作为药物溶解度、渗透性、稳定性、蛋白质结合和血浆 PK 参数的可接受标准,还对先导化合物的药物样特性进行了广泛优化。尽管在过去的几十年里,在药物开发过程的各个方面都做出了巨大的努力,但药物开发的成功率仍然保持在 10%-15%5-7。持续的高失败率引发了一个问题,即尽管经过验证的分子靶点,药物优化的某些方面是否被忽视。
3.2.2. 在药物优化中,忽视了疾病靶向组织与健康组织中药物暴露/选择性的平衡,这可能会误导候选药物的选择并改变临床剂量/功效/毒性的平衡
在临床药物开发中,需要在临床剂量、疗效和毒性之间实现微妙的平衡,以优化患者的受益/风险比。理想的候选药物应具有高效力和特异性,抑制其分子靶点而不会产生脱靶应,在疾病靶向组织中的高药物暴露可在最佳剂量(理想情况下为低剂量)达到够的疗效,以及健康组织中的最小药物暴露可在最佳剂量(即使是高剂量)下避免毒性。然而,当前药物优化过程可能过度强调了使用SAR研究的效力/特异性,而忽视了疾病靶向组织与健康组织中药物暴露/选择性的平衡,这可能误导了候选药物的选择,打破了临床剂量/疗效/毒性之间的平衡,导致高临床失败率。如果给予足够的剂量,使用当前药物优化标准选择的候选药物通常在体外和动物模型中显示出良好的疗效。然而,一旦选择了候选药物并进入临床试验,最重要的问题之一是当达到足够的疗效时患者是否能耐受毒性。由于缺乏疗效而导致的临床药物开发失败,往往并不意味着候选药物不起作用,但很可能是因为这些药物即使在健康器官中最大耐受剂量(MTD)下显示出毒性,也无法在疾病靶向器官中显示出令人满意的疗效。这些药物在高于 MTD 的剂量下肯定会在疾病靶向器官中显示出疗效,但患者无法耐受健康器官中的高剂量。另一方面,由于无法控制的毒性而导致临床药物开发失败的原因很可能是因为它在药物对疾病靶向器官产生任何功效之前,即使在低剂量下也对正常健康器官显示出毒性。因此,成功/临床药物开发的失败取决于临床剂量、疾病靶向器官的疗效和正常健康器官的毒性之间的微妙平衡。因此,重要的是测试候选药物是否可以在疾病靶向器官中达到足够的暴露,而不会在健康的正常器官中暴露过多的药物。然而,在药物优化过程中没有考虑这些方面,这可能会误导先导候选药物的选择,影响临床剂量/疗效/毒性的平衡。这可能是药物优化过程中被忽视的属性之一,尽管分子靶点经过验证,但仍导致临床药物开发的高失败率。幸运的是,在针对中枢神经系统(CNS)的药物优化过程中,除了优化 SAR 和类似药物的特性外,大脑中的药物暴露或药物穿过血脑屏障(BBB)的能力始终是候选药物选择的选择标准52,54-57,67。人们普遍认为,如果候选药物难以穿透血脑屏障到达脑组织,则无法达到足够的疗效,应在药物开发早期终止。然而,其他治疗领域的药物优化很少采用标准来确保药物暴露于疾病靶器官与正常器官中。对于抗癌药物的发现,靶点参与确实经常在异种移植模型中进行评估,或者通常在人类肿瘤切除术中进行评估58-62,68-71。然而,由于高血浆药物暴露足以实现高肿瘤暴露,因此很少评估药物分子的结构修饰如何改变肿瘤与正常组织中的药物暴露。在抗癌药物的临床试验中,由于在临床研究中很容易获得外周血单个核细胞,因此通常在外周血单个核细胞(PBMC)中监测靶点参与情况以进行临床剂量选择。然而,外周血单个核细胞中的靶向参与是一种较差的替代生物标记物,在低剂量时很容易饱和,而肿瘤组织中的靶向参与则勉强够用。人们普遍认为,基于外周血单个核细胞靶向参与的血浆药物暴露的剂量优化只是实体肿瘤剂量优化的参考和起点,实现最大肿瘤抑制或消退的最终剂量远高于在血液中产生最大靶向参与的剂量。一旦在临床研究中进行剂量递增,临床剂量/疗效/毒性的次优平衡可能导致临床开发失败。例如,对美国食品和药物管理局(FDA)批准的EGFR抑制剂(吉非替布、拉帕替尼、厄洛替尼和凡德他尼)的分析表明,经过某些修饰的类似药效团具有不同的适应症,可治疗具有不同抗癌功效的不同癌症类型72-75。然而,这些EGFR抑制剂的SAR和类药物特性不能完全解释其独特的临床抗癌效果和毒性。除SAR和药物样特性外,结构修饰可能会显著影响其在不同肿瘤类型中的药物暴露,这可能显著影响其抗癌疗效(未公布的数据)。同样,一些成功的BTK抑制剂,如伊布替尼、阿卡替尼和泽布替尼显示出不同的疗效/毒性特征76,77,而司培替尼在早期临床试验78中失败。除了药物特异性/效力和类药物特性外,它们在疾病靶器官与正常器官中的组织暴露也可能有助于其疗效/毒性,这需要进一步研究。此外,对FDA批准或临床失败的选择性雌激素受体调节剂(SERMs)的分析也表明,轻微的结构修改可能会改变临床剂量、临床开发之间的平衡79-83。例如,已经为SERMs临床开发进行了600多个临床试验,其中11个SERMs已获得批准,许多SERMs在临床研究中失败81-83。一些SERMs具有非常相似的结构,只有非常轻微的修改,但它们在治疗乳腺癌、骨质疏松症和更年期症状的各种适应症中显示出明显的疗效。它们的SAR和类药物特性不能完全解释其临床剂量、疗效和毒性之间的差异79-83。先前的研究表明,轻微的结构修饰可能显著改变不同组织(如肿瘤、脂肪垫和骨骼)中的药物暴露和药物选择性,从而影响其临床剂量、疗效和毒性40。此外,最新例子用于治疗 COVID-19 的抗病毒药物瑞德西韦,也证明了药物暴露在疾病靶向组织与正常组织中对于剂量/功效/毒性的微妙平衡的重要性。尽管在体外对严重急性呼吸综合征冠状病毒 2 (SARS-CoV-2) 具有良好的体外活性,但瑞德西韦在治疗 COVID-19 方面的临床疗效非常有限84,85。在100 mg的剂量下,瑞德西韦及其活性代谢物的暴露/选择性可能太低,无法在肺部实现出色的杀死 SARS-CoV-2 病毒的功效,但在肾脏中却太高而无法引起毒性86。此外,许多方法已被用于估计肺中抗感染药物的药物浓度,以研究它们对疾病调节的功效37, 53。这些例子清楚地表明,候选药物的选择和临床剂量/疗效/毒性的微妙平衡不仅由SAR和类药物特性决定,而且还受到疾病靶向组织与正常健康器官中药物暴露平衡的影响,这在药物优化和临床研究中经常被忽视。4.如何改进药物优化以选择更好的候选药物,平衡临床剂量/疗效/毒性,并提高临床药物开发的成功率?
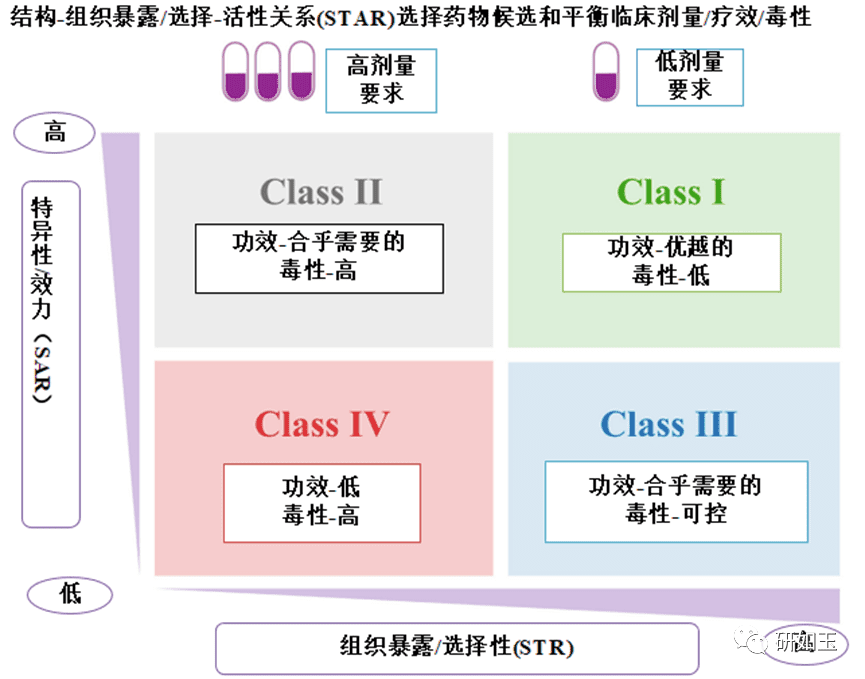
临床药物开发的成功将取决于最佳候选药物的选择以及临床剂量/功效/毒性之间的微妙平衡,以及真正的靶点验证。候选药物在人体试验中的临床剂量/功效/毒性的平衡不仅取决于其抑制其分子靶标的效力/特异性(通过 SAR 研究),而且还取决于其在疾病靶向器官中的暴露/选择性与其他器官的暴露/选择性。正常器官(通过结构–组织暴露/选择性关系,STR)。我们提出了一种结构–组织暴露/选择性活性关系(STAR)系统来改进药物优化过程。STAR系统根据三个方面将候选药物分为四个不同的类别(I‒IV):使用IC50或Ki(高或低)的SAR研究抑制分子靶的药物效力/选择性;使用STR研究的药物组织暴露/选择性(高或低);平衡临床疗效/毒性的剂量要求(高或低)。四种不同类别的候选药物(I‒IV类)需要不同的策略来选择先导候选药物,优化临床剂量,并平衡临床疗效/毒性。STAR的成功应用将提高四类不同候选药物的药物优化和临床研究效率,以提高临床药物开发的成功率(图2)。
图 2结构–组织选择性/暴露活性关系 (STAR) 选择更好的候选药物并平衡临床剂量/功效/毒性,以改进药物优化,以实现成功的临床药物开发。
在这个 STAR 系统中,SAR 探索改变化合物对分子靶标的结合亲和力和特异性的结构修饰。使用 SAR 可以优化药物效力和特异性,以获得在低 nmol/L 或 pmol/L 范围内具有低 Ki 或 IC50 的化合物,从而获得更好的疗效和更低的毒性6,7,87。SAR 研究已经在当前的药物优化过程中得到了很好的确立和验证。此外,STR研究了改变疾病靶向组织与正常健康器官中药物暴露/选择性的结构修饰,这不仅影响药物优化过程中的先导化合物选择,而且决定了临床剂量/功效/毒性之间的平衡在人体试验中40,88。然而,STR研究在药物优化过程和临床试验设计方面尚未得到很好的确立。此外,临床疗效/毒性的平衡始终是剂量依赖性的。为了成功开发临床药物,优化患者的受益/风险比,始终需要在临床剂量/疗效/毒性之间实现微妙的平衡。4.1 Class I—I 类
I 类候选药物对其分子靶点具有高特异性/效力(通过 SAR 研究,Ki 几百 nmol/L),并且在疾病靶向组织中具有高暴露/选择性(通过 STR 研究)。 I类候选药物需要低剂量才能达到足够的疗效。健康器官中的毒性较低,这得益于疾病组织的高选择性和正常器官中的低组织暴露/选择性。 I 类药物在血浆中的药物暴露量可能高或低。 I 类候选药物是最理想的,在临床剂量/功效/毒性的临床试验中具有最高的成功率。例如,以下成功获批的药物很可能是I类药物,如抗病毒药物索非布韦(NS5B聚合酶抑制剂)、胆固醇治疗药物阿托伐他汀(HMG-CoA还原酶抑制剂)、勃起功能障碍治疗药物西地那非(PDE5抑制剂)和抗癌剂阿卡替尼(BTK抑制剂)。值得注意的是,与血浆中的药物暴露相比,一些I类药物在疾病靶向组织和一些正常器官(与毒性有关)中也可能具有高暴露/选择性,这可能兼具高疗效和高不良反应发生率。不良事件。以下药物很可能是这些类型的例子,例如抗癌药凡德他尼(VEGFR/RET/EGFR 抑制剂)、他莫昔芬(SERM)、泊马度胺(E3 连接酶抑制剂)和阿霉素(拓扑异构酶I 抑制剂)。4.2 Class II—II类
II 类候选药物对其分子靶点具有高特异性/效力(通过 SAR 研究,Ki 几百 nmol/L)和在疾病靶向组织中的低组织暴露/选择性(通过 STR 研究)。为了在疾病靶向组织中获得足够的疗效,II 类候选药物通常需要高剂量,因为它们在疾病靶向器官中的暴露/选择性低。然而,由于在病变组织中的高剂量和低组织暴露/选择性(但在正常器官中的高组织暴露/选择性),毒性可能变得难以控制。目前,大多数药物优化工作都集中在这类II类化合物上,以提高其特异性/效价(Ki)。先导化合物通常优化为具有低nmol/L甚至pmol/L IC50或Ki。在先导候选药物选择过程中,通常选择血浆暴露量高、血浆PK参数较好的化合物进行临床研究39,89。然而,血浆中的体内药物浓度通常需要达到mmol/L才能达到足够的疗效,尤其是对于抗癌药物。人们会想知道为什么体外IC50和体内EC50之间存在很大差异。其中一个重要因素可能是由于疾病靶向组织中药物暴露/选择性较低。在这种情况下,血浆中的高药物暴露可能会误导候选药物的选择和临床剂量/疗效/毒性的优化。由于血浆中的药物暴露取决于消除(清除)和分布(药物在不同器官中)过程90,因此高血浆暴露的候选药物可能归因于低消除或低组织分布。在疾病靶组织中,血浆暴露量高但组织暴露量低的药物可能不是首选药物,因为低靶组织暴露可能导致疗效不足。在这种情况下,通常使用高剂量来达到足够的疗效,但如此高的剂量可能会导致其他重要器官的高毒性91。如果候选药物在疾病靶器官中具有较低的组织选择性和暴露,但在其他重要健康器官中具有较高的组织选择性和暴露,则其治疗窗口可能较窄,并且可能由于高毒性而无法达到其治疗浓度。此外,如果候选药物在疾病靶向和健康生命或器官中的选择性和暴露度较低,即使在I期研究中药物血浆暴露量较高,也可能不会有安全问题,但在II/III期研究中可能达不到预期的疗效。不幸的是,我们经常观察到此类先导化合物,它们具有高效价/特异性、低组织暴露/选择性、高血浆暴露、良好的安全性,在I期研究中大放异彩,但在随后的III期研究中由于缺乏疗效而失败。因此,应谨慎评估II类候选药物的适应症、剂量优化以及临床疗效和毒性的平衡。有几种药物可能是II类药物,例如抗癌药物伊布替尼(成功的BTK抑制剂)、泽布替尼(失败的BTK抑制剂)和菲达替尼(成功的JAK2抑制剂)。如果抗病毒药物瑞德西韦(RdRp抑制剂)的抗病毒活性被认为很高(Calu-3细胞中低nmol/L的IC50),那么它也可能是II类药物。然而,如果瑞德西韦的抗病毒活性被认为较低(在Vera细胞中的IC50为mmol/L),则可能被归类为如下所述的IV类药物。4.3 Class III—III 类
III 类候选药物对其分子靶点具有低但合乎需要的特异性/效力(SAR 研究为数百 nmol/L ),但在疾病靶向组织中具有高组织暴露/选择性(STR 研究)。 III类候选药物需要中低剂量才能达到足够的疗效。由于低至中等剂量、疾病靶向器官中的高暴露/选择性和正常器官中的低暴露/选择性,毒性是可控的。目前,III 类化合物在血浆中的暴露量可能很低,很可能被忽视。由于血浆药物暴露量低,它们通常在药物优化过程的早期阶段被终止。然而,低血浆暴露也可能是由于高组织分布,特别是当候选药物具有良好的体内稳定性和相对合理的生物利用度时。此类化合物可能具有较高的靶组织暴露量,这有利于在低剂量下获得更好的疗效,而低剂量也可以最大限度地减少对健康器官的毒性。这些III类候选药物可以平衡临床剂量/功效/毒性,从而提高从I期到III期临床试验的成功率。然而,这些类型的化合物中有很大一部分可能在进入临床试验之前就被错误终止。值得注意的是,III类化合物的特异性和效力仍然需要IC50 ,因为无论组织暴露/选择性如何,IC50 >数mmol/L的化合物可能无法达到足够的疗效。市场上属于 III 类药物的药物很少,因为许多 III 类候选药物可能已在早期药物发现过程中终止。抗癌药沙利度胺(E3 连接酶抑制剂)可能属于 III 类药物。4.4 Class IV—IV 类
IV类候选药物对其分子靶标的特异性/效力低(通过 SAR 研究,Ki > 1到数mmol/L)和在疾病靶组织中的低暴露/选择性(通过 STR 研究)。IV类候选药物通常需要高剂量才能达到一定程度的功效,但仍然不足。由于在疾病靶向器官中的高剂量和低组织暴露/选择性,但在正常器官中的高组织暴露/选择性,毒性将变得难以控制。IV类候选药物是最不受欢迎的,应在药物优化过程的早期阶段终止。值得注意的是,在进入临床试验期间,无论组织暴露/选择性如何,都应谨慎评估具有低效力/特异性(Ki > 1至数mmol/L)的候选药物。大多数IV类候选药物可能在临床开发中失败,成功的例子非常有限。值得注意的是,由于STR目前未被用作药物优化过程,因此每个类别(IeIV)中的药物示例是基于其血浆PK和临床剂量/疗效/毒性进行估计的,而没有实验数据。如下所述,需要更详细的研究来根据它们的组织暴露/选择性将这些药物准确地分为四个不同的类别(I‒IV)。值得注意的是,真正的靶点验证对于临床药物开发的成功同样重要。此外,目前成功的策略是有效的,并且有充分的理由克服90%临床开发失败的四个可能原因。被忽视的STAR是临床药物开发90%失败率需要考虑的主要因素之一,但不是唯一因素。STAR的应用将提高四类不同候选药物的药物优化和临床研究效率,从而提高成功率。然而,单独应用STAR并不能保证临床药物开发的成功率达到90%-100%,但它可以显著提高成功率。成功率从10%-15%提高到30%-40%将对整个药物开发产生重大影响。5.如何在药物优化过程中实施 STAR 及未来展望
5.1 SAR和STR的测量
SAR 研究已广泛应用于药物优化过程,使用计算辅助药物设计和体外筛选,通过测量化合物与其分子靶点结合的 IC50 或基于蛋白质测定或基于细胞的测定Ki (nmol/L) 。之前已被广泛讨论16-20,未包含在本评论中。靶点验证也已在之前也进行了广泛讨论8-12,未包含在本评论中。然而,STR研究很少被用作当前药物优化中的药物优化标准来选择先导候选药物并平衡临床剂量/疗效/毒性。定义高或低组织暴露/选择性的临界值尚未定义,需要进一步研究。在实验上,STR 可以使用药物组织暴露 (AUC)、分配系数Kp、所有器官的组织选择性 (%) 以及与毒性相关的疾病靶向组织与正常重要器官的 AUC 比率来测量。值得注意的是,组织暴露/选择性概念不同于传统药代动力学中的分布容积 (V)。药物在每个器官中的组织暴露/选择性将由药物在每个器官中的吸收/生物利用度、分布、蛋白质/组织结合、代谢决定。每个器官的组织暴露/选择性将不同于另一个器官。传统的分布容积是一个数学术语,用于描述药物分子在一般组织中分布的难易程度,但它无法区分哪个组织高或低。5.2 STR 的组织暴露/选择性测量
药物组织暴露(AUC)描述了某些组织中累积的总药物(结合和游离)量,该量由血浆和组织中的药物暴露(AUC)决定,使用分配系数(Kp)92,如等式(1)所示:组织中的药物暴露(AUC)=血浆中的药物暴露(AUC)×Kp其中,血浆和组织中的药物暴露可使用总药物浓度与时间曲线计算,Kp值可通过组织与血浆中的总药物浓度(Ctissue/Cplasma)来计算或组织与血浆中总药物的AUC(AUCtissue/AUCplasma)92来计算。药物组织选择性影响药物的治疗窗口以平衡剂量、疗效和毒性。可以使用两个方程式来描述,如等式(2) 和 (3),视各种实验目的和条件而定。药物组织选择性可以用药物在与疗效或毒性有关的某些组织中的总药物浓度或暴露量占所有组织总药物浓度或暴露量的比例来描述,,如等式(2)所示:药物组织选择性=Ctissue/ΣCtissueorAUCtissue/ΣAUCtissue (2)其中,Ctissue或AUCtissue的总和是组织中的总药物浓度或AUC。等式(2)适用于毒性特征未知的候选药物。测定所有主要组织中的药物选择性可以提供有关候选药物潜在毒性的信息。然而,测定所有主要组织中的药物组织总暴露量既耗时又费力;因此,它仅适用于从少量候选药物中筛选。因此,药物组织选择性也可以通过与药物疗效相关的疾病靶向组织中的总药物浓度或暴露量与与毒性相关的健康重要器官中的总药物浓度或暴露量的比率来描述,如等式(3)所示:药物组织选择性比=Cteff/CtoxorAUCeff/AUCtox (3)其中,Ceff或AUCeff是组织中与疗效相关的的药物浓度或AUC,Ctox或AUCtox是组织中与毒性相关的的药物浓度或AUC。当清楚证明候选药物的主要或致命毒性并与一个或两个组织相关时,可以使用等式(3)。等式(3)不需要测量所有组织浓度,这更省时省力。5.3 STR总药物浓度/暴露与游离药物浓度/暴露的测定
根据传统的小分子“游离药物假说”,上述计算中是否应该使用总药物或游离药物的浓度或暴露量来进行先导候选药物的选择是有争议的。 “游离药物假说”认为血浆药物总浓度包括游离非结合药物和血浆蛋白结合药物之间的平衡41,93;只有游离的非结合药物(而非蛋白质结合药物)才能分布到其他组织,而蛋白质结合药物在血浆中作为储存器释放游离药物41,93;组织中的总药物浓度还包括游离的非结合药物和结合药物;只有游离的非结合药物才能与其分子靶点相互作用以发挥药理功能41,93;在稳态下,组织中的游离药物浓度与血浆中的游离药物浓度相似或相等41,42,因此,血浆中的药物浓度或暴露量可用于预测靶组织中的药理作用。尽管只有游离药物才能与其分子靶点发挥药理作用,而结合药物可以作为释放游离药物发挥药理作用的储存器是正确的,但基于以下原因假设只有游离药物才能分配到其他组织是错误的: (1) 只有游离药物才能分配到组织中的假设完全忽略了血浆蛋白本身(如白蛋白)从体循环到组织的运输。事实上,白蛋白通过 FcRn 和其他主动转运过程45从血浆主动转运到细胞外基质,再转运到组织,将脂肪酸或药物运送到不同的组织。先前的研究发现,白蛋白结合小分子(酪氨酸激酶抑制剂)与血管和组织中的白蛋白结合蛋白相互作用,如 gp18、gp30、gp60/清蛋白激活蛋白以及分泌型酸性蛋白和富含半胱氨酸蛋白 (SPARC),其介导这些小分子在正常组织中的组织积累,这与它们的毒性有关46。与正常组织相比,与蛋白质结合的药物在肿瘤组织中的积累也可能更高40。(2)“游离药物假说”在下列情况下是不正确的,因为血浆和组织之间发生不对称的游离药物分布:参与药物吸收和排出的吸收和流出药物转运体(例如,肝脏和大脑)42-44,49;电离药物受 pH 梯度和“溶酶体捕获”效应影响42,50;与靶点共价结合的药物42;前药和蛋白质降解剂42。众所周知,游离药物假说不适用于抗体–药物偶联物、纳米药物、核酸42和局部给药的药物(例如吸入)37,48,94,这些不包括在本综述中。因此,“游离药物假说”仅在药物被动地从血浆扩散到其靶点并且没有迅速清除的某些有限情况下是正确的,但在许多其他情况下是不正确的42。在药物优化过程中,游离药物组分在血浆和组织中都是非常重要的参数,因为只有游离药物才能与其分子靶点结合以获得药理功能。然而,在药物优化过程中仅通过测定血浆蛋白结合来过分强调血浆中的游离药物部分可能会造成误导。由于游离非结合药物和蛋白结合药物在正常组织和疾病靶向组织中都存在平衡和运输,我们建议使用疾病靶向器官与正常器官的总药物暴露/选择性51-53。以往的临床前和临床研究也直接使用总组织暴露或 Kp(总药物在组织/血浆中的比率)来筛选候选药物,并评估剂量依赖性疗效/毒性54-64。阿斯利康临床开发组建立了转化药代动力学和药效学 (PK/PD) 模型,这表明对于临床 PK/PD 关系,总药物水平优于游离药物水平48,64。组织中的游离药物浓度可能仍然非常有用,但很难测量。它在先导药物候选选择上的应用仍有争议。测定组织中游离药物(fu)的方法有多种,其中平衡透析是最常用的方法95。然而,目前大多数测定fu的方法使用的是组织匀浆,这种方法破坏了所有亚细胞结构,并且不能真正代表作用部位组织中的游离药物浓度42,52。5.4 开展体外高通量筛选以研究STAR
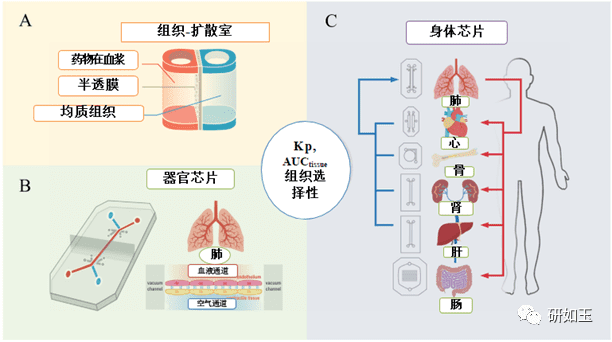
SAR 的高通量研究已在药物优化过程中成功实施 8,10,16-20,因此在此不再赘述。然而,仍然缺乏高通量 STR 方法。未来,除了 SAR 研究,在优化过程的早期阶段,开发体外筛选工具用于筛选 STR 将是理想的。扩散室可以提供一种简单且高通量的方法来筛选组织暴露和候选药物的选择性(图 3A)。随着时间的推移,可以测定从血浆室(模拟全身循环中的药物)扩散到均质组织室(模拟不同的组织,如肝脏、肾脏、心脏等)的药物量。血浆分配系数 (Kp) 可以用不同时间点的组织室/血浆室的总药物浓度或 AUC 比率来计算。同时,药物在不同组织中的总药物暴露量和组织选择性可以使用等式(1)–(3)计算,并通过与动物模型体内组织暴露和选择性的相关性来证实。
图 3 用于研究结构组织选择性/暴露活性关系 (STR) 的体外高通量筛选工具。 (A) 组织扩散室可以为研究结构–组织暴露/选择性关系 (STR) 提供简单且高通量的筛选。 (B) 单器官芯片,和 (C) 身体芯片集成多个器官单元以研究不同器官中的 STR。
器官芯片技术的出现也可能为STR提供更好的HTS(图3B和C)。器官芯片技术可以结合人体生理条件,有助于药物组织暴露/选择性。各种单器官芯片通过器官(如血管网络、肺和肠道)的多细胞血管或上皮界面提供组织屏障,决定药物在某些组织中的渗透。一些器官芯片也可以提供组织水平的组织,例如实质细胞(例如肝脏、心脏、骨骼肌和肿瘤),这有助于药物与特定组织的结合96,97。可以开发多器官芯片或“身体芯片”来集成模拟整个人体的多个器官单元,并确定不同器官组织中的STR 98。5.5 开发人工智能(AI)辅助计算模型对STAR进行评估
人工智能辅助计算工具已成功用于分子靶标的三维蛋白质结构预测、药物分子抑制分子靶标的设计以及药物分子与其靶标相互作用的SAR研究15,99-101。虽然已经有人尝试使用人工智能和机器学习技术来利用化合物的结构信息来预测药物样特性和血浆PK102,103,但这些新技术尚未成功用于STR的研究,理想的做法是使用人工智能和机器学习进行结构表示(分子图表示法、线性表示法或化学描述符),在先导化合物选择和临床试验设计中预测STR,并平衡剂量效应毒性。例如,可以使用化学信息学对先导化合物的物理化学描述符进行计算分析。这些描述符可以包括但不限于结构键、散列指纹;自相关、电荷、logP、蛋白质结合、折射率、成分、拓扑和连接性描述符、来自MOE(分子操作环境)的复合描述符和Kappa形状指数。基于人工智能的计算模式可用于分析化学描述符与组织暴露、组织/血浆分配系数和组织选择性之间的关系,用于具有体外和体内STR数据40的一组选定化合物。一旦通过AI辅助计算建模建立了足够数量的化合物的信息,对于任何新设计的化合物,SAR 和 STR 的预测将在合成前使用基于 AI 的计算分析进行,这可能会减少药物优化过程中的工作量。此外,FDA 批准药物的基于 AI 的计算建模所需的信息已包含在 FDA 数据库的 NDA 应用程序包中,例如理化性质、不同组织中的碳质量平衡研究14、临床疗效以及临床前和临床毒性。如果可以收集这些数据,则可以使用人工智能辅助计算分析来预测STR与临床剂量、疗效、不良事件和毒性的关系。然而,这项工作需要学术界、FDA和制药行业之间的合作,以开发和验证这些人工智能辅助计算工具。传统上,基于生理的药代动力学 (PBPK) 建模已用于根据血液(不仅是血浆)中的总药物浓度预测药物组织总暴露量,这可以从动物模型扩展到人类。然而,PBPK 建模依赖于来自临床前动物模型的大量组织浓度数据,这些数据是劳动密集型的。如果可以使用扩散室或器官芯片的体外筛选方法来预测组织暴露/选择性,则可以基于体外筛选数据建立 PBPK,这可能会提高STR在先导化合物选择和临床试验设计中的预测能力和实际应用。 PBPK 建模结合了每个器官中的所有生理参数,以预测所有组织中的药物浓度随时间变化37,104。目前,PBPK 模型已被整合到药物开发过程和监管提交文件中,其主要目的是定性和定量地预测药物相互作用并将 PK 特性扩展到人体,以支持儿科和首次人体试验中的初始剂量选择37,105。然而,使用 PBPK 建模预测临床组织 PK 曲线有挑战性,原因如下:(1)基于化合物特异性临床前数据构建的PBPK模型通常无法随着临床观察而细化和更新,其中人体组织暴露难以检测; (2) 化合物的轻微结构修饰可能会显著改变不同器官中的组织暴露/选择性,而传统的 PBPK 无法在先导化合物选择或临床试验设计的实时决策中实际执行。一些初步研究已经使用PBPK模型在12种吸入性支气管扩张药的动物物种和人类之间翻译药物的PK,用于研究药物对肺功能的剂量依赖性临床效应37,64。5.6 临床前动物模型研究STAR
目前,根据SAR研究选择的几种先导化合物在疾病动物模型中进行常规测试其疗效。针对一个分子靶标测试5-10个化合物是可行的,本文不再赘述。然而,用于STR研究的化合物的临床前动物试验需要在所有劳动密集型组织中测量药物暴露/选择性。因此,在STR研究中测试数百种化合物是不可行的。因此,重要的是决定应该实施 STR 研究的药物优化过程的哪一步。在此,我们提出了一种可行的早期药物优化策略,利用STAR选择用于临床研究先导化合物(图4)。一般来说,在单个药物靶点的药物优化过程中,大约会合成200-400个化合物用于SAR研究。在筛选这些化合物的体外特异性和效力的体外活性后,数十种化合物进行体外ADME筛选(溶解度、渗透性、稳定性和蛋白结合),以根据血浆 PK 曲线选择一些化合物进行体内药代动力学研究。除了功效测试外,还应进一步测试几种具有可接受的血浆 PK 曲线的化合物以进行 STR 研究,以确定它们在临床前动物模型中的组织暴露和选择性。应综合考虑体外和体内活性(SAR)、体外ADME、体内血浆PK、体内组织暴露和选择性(STR)等因素,选择最佳先导化合物用于临床研究,提高药物开发的成功率。
图 4 当前药物优化过程中先导化合物选择的结构–组织选择性/暴露活性关系 (STAR) 的实施。
5.7 开发人体无创成像技术以在临床试验中评估STAR
理想情况下,体内成像技术将非常有助于在人体临床试验中研究STAR,以提高药物开发的成功率。虽然成像模式已用于药物开发的临床试验,但目前还没有合适的成像技术用于临床试验中研究STAR。动态位置发射断层扫描(PET)成像可用于可视化放射性标记的化合物,用于体内靶点接合或观察化合物组织暴露,两者均可用于研究STR。例如,靶参与的定量评估提供了一种可视化药物分布的潜在方法,并为剂量选择提供了直接证据,以在靶器官中实现充分的药物暴露,确保药理活性88。PET成像已用于临床,使用放射性标记的神经激肽1(NK1)受体拮抗剂评估组织中的靶点参与,这有助于临床试验的决策。临床PET成像使用[18F]SPARQ研究受体占有率,结果表明拮抗剂对NK1受体的高占有率并未转化为治疗效果。PET成像研究证实,缺乏疗效是由于药理机制的假设无效,而不是由于大脑中药物暴露不足,这导致决定停止使用NK1拮抗剂进行抗抑郁或焦虑治疗106,107。相反,阿瑞匹坦(一种选择性NK1拮抗剂)在脑中的充分药物暴露后来被批准用于预防急性和延迟化疗引起的恶心和呕吐108。PET成像研究有助于优化剂量,以表现出完全的中枢神经系统靶点参与,从而在治疗癌症患者时获得足够的疗效并最大限度地减少药物的相互作用。然而,最常用的正电子发射放射性同位素衰变很快,半衰期相对较短(例如,C-11为20分钟,F-18为110分钟),这仅允许短期检测药物组织暴露/选择性和靶参与109。尽管PET成像中的F-18标记化合物的半衰期稍长(110分钟),但如之前的研究40所示,F-18标记化合物可能会改变不同器官中的药物暴露和选择性。化合物的C-11标记可能对药物组织暴露/选择性影响最小,但其半衰期仅为11分钟。C-14标记化合物广泛用于动物模型和人体的质量平衡研究。C-14标记化合物在动物模型中的分布研究可能为基于组织暴露/选择性的先导化合物选择提供有用信息,但它仅用于人体的质量平衡研究,而不知道组织暴露/选择性110。其他体内成像方法,如磁共振成像,只能显示人体解剖结构,而不能以足够的灵敏度和特异性显示体内药物分子。质谱成像(MSI)目前仅用于观察离体组织切片中的药物分子,而MSI的手持式探针正在开发过程中;因此,它还不能检测或想象人体内的药物分子。显然,未来需要更多用于研究 STAR 的成像方式来选择候选药物并平衡临床剂量/功效/毒性。6 结论
在过去的几十年里,90%的临床药物开发在临床I、II、III期临床研究和药物批准过程中失败,原因可能有四个:缺乏临床疗效、毒性难以控制、不良的类药性特性、缺乏商业需求和糟糕的战略规划。尽管许多成功的策略都被正确实施,克服了 90% 临床开发失败的四种可能原因,但在过去的几十年里,临床药物开发的成功率仍然保持在 10%-15%。如此高的失败率引发了一个问题,即药物开发的某些方面是否被忽视了?一方面,真正的靶点验证,确认分子靶点是人类疾病的原因和药物的预期靶点,对临床药物开发的成功仍然具有挑战性。另一方面,当前的药物优化可能过分强调了一个方面,而忽略了可能误导候选药物选择和临床剂量/疗效/毒性失衡的其他方面。在临床药物开发中,为了在临床剂量、疗效和毒性之间实现微妙的平衡,以优化患者的受益/风险比,理想的候选药物应具有高效力和特异性,以抑制其分子靶点而不产生脱靶效应,疾病靶标组织中的高药物暴露以在最佳剂量(理想的低剂量)下获得足够的疗效,以及在健康组织中的最小药物暴露以避免最佳剂量(即使在高剂量)下的毒性。在临床试验中,候选药物的临床剂量/疗效/毒性的微妙平衡不仅取决于它们抑制其分子靶标的效力/特异性(通过SAR研究),还取决于它们在疾病靶标组织和正常组织中的组织暴露/选择性(通过STR研究)。然而,目前的药物优化过程通过SAR研究过分强调药物的效力/特异性,而通过STR研究忽略了药物在疾病靶向组织与正常组织中的组织暴露/选择性,这可能会误导药物候选选择,影响临床剂量优化,以及打破了临床疗效和毒性的平衡。我们提出了一个 STAR 系统来改进药物优化过程,该系统基于三个方面将候选药物分为四个不同的类别:药物效力/特异性(高或低)、药物组织暴露/选择性(高或低)和平衡临床疗效/毒性所需的剂量(高或低)。四种不同类别的候选药物(I-IV 类)需要不同的策略来选择先导候选药物、优化临床剂量和平衡临床疗效/毒性。在这个 STAR 系统中,I 类候选药物具有高特异性/效力和高暴露/选择性,需要低剂量才能达到平衡的疗效/安全性,是最理想的,成功率高。 II类候选药物具有高特异性/效力和低组织暴露/选择性,需要高剂量来获得足够的疗效,但可能具有无法控制的毒性。 II 类候选药物需要谨慎评估,以平衡临床剂量/疗效/毒性。 III 类候选药物具有相对较低但足够的特异性/效力,但组织暴露/选择性较高,这可能需要低至中等剂量才能达到足够的疗效和可控的毒性。 III类候选药物可能具有较高的临床成功率,但由于在药物发现的早期阶段血浆药物暴露不良而经常被忽视。 IV类候选药物特异性/效力低、暴露/选择性低,往往需要大剂量、疗效不佳、毒性大,应尽早终止。未来,STAR系统可以通过人工智能辅助计算建模、体外筛选、体内测试和无创成像技术进行改进。 STAR的应用将提高四种不同类别候选药物的药物优化和临床研究效率,从而提高临床药物开发的成功率。作者贡献
孙杜欣(音译)萌生了这个想法,写下了手稿。魏高(音译)写了手稿。胡鸿祥(音译)修改稿件,设计框架。 Simon 周修改了想法和手稿。所有作者都已阅读并批准了最终手稿。利益冲突
作者没有需要声明的利益冲突。 Simon 周是百时美施贵宝公司的员工。